

Heterozygous BTNL8 variants in individuals with multisystem inflammatory syndrome in children (MIS-C)
- PMID: 39576310
- PMCID: PMC11586762
- DOI: 10.1084/jem.20240699
Heterozygous BTNL8 variants in individuals with multisystem inflammatory syndrome in children (MIS-C)
Abstract
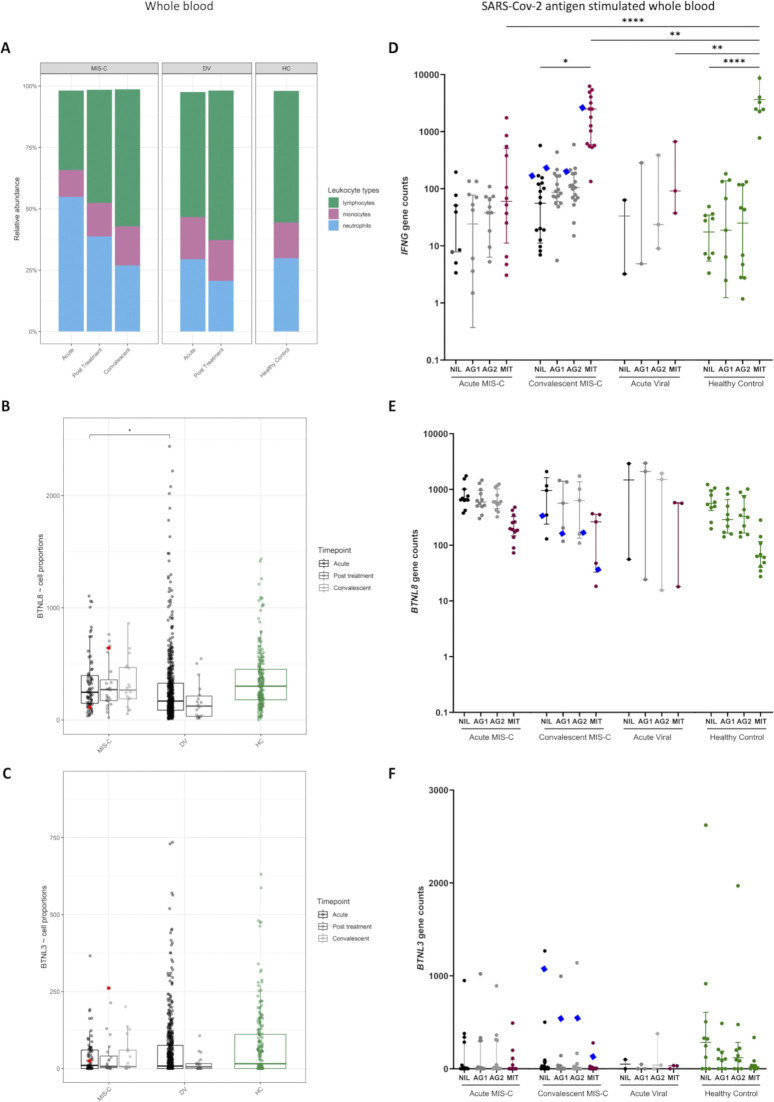
Multisystem inflammatory syndrome in children (MIS-C) is a rare condition following SARS-CoV-2 infection associated with intestinal manifestations. Genetic predisposition, including inborn errors of the OAS-RNAseL pathway, has been reported. We sequenced 154 MIS-C patients and utilized a novel statistical framework of gene burden analysis, "burdenMC," which identified an enrichment for rare predicted-deleterious variants in BTNL8 (OR = 4.2, 95% CI: 3.5-5.3, P < 10-6). BTNL8 encodes an intestinal epithelial regulator of Vγ4+γδ T cells implicated in regulating gut homeostasis. Enrichment was exclusive to MIS-C, being absent in patients with COVID-19 or bacterial disease. Using an available functional test for BTNL8, rare variants from a larger cohort of MIS-C patients (n = 835) were tested which identified eight variants in 18 patients (2.2%) with impaired engagement of Vγ4+γδ T cells. Most of these variants were in the B30.2 domain of BTNL8 implicated in sensing epithelial cell status. These findings were associated with altered intestinal permeability, suggesting a possible link between disrupted gut homeostasis and MIS-C-associated enteropathy triggered by SARS-CoV-2.
© 2024 Bellos et al.
Conflict of interest statement
Disclosures: A.H. Tremoulet reported non-financial support from Janssen Pharmaceuticals outside the submitted work. C.-E. Luyt reported personal fees from Advanzpharma, grants from Merck, and non-financial support from Pfizer outside the submitted work. A.C. Hayday reported grants from Takeda Pharmaceuticals, and personal fees from ImmunoQure AG, Prokarium, and TransImmune AG outside the submitted work; in addition, A.C. Hayday had a patent to US20210246187A1 pending (Takeda). No other disclosures were reported.
Figures










References
-
- Abolhassani, H., Delavari S., Landegren N., Shokri S., Bastard P., Du L., Zuo F., Hajebi R., Abolnezhadian F., Iranparast S., et al. 2022. Genetic and immunologic evaluation of children with inborn errors of immunity and severe or critical COVID-19. J. Allergy Clin. Immunol. 150:1059–1073. 10.1016/j.jaci.2022.09.005 - DOI - PMC - PubMed
-
- Assante, G., Tourna A., Carpani R., Ferrari F., Prati D., Peyvandi F., Blasi F., Bandera A., Le Guennec A., Chokshi S., et al. 2022. Reduced circulating FABP2 in patients with moderate to severe COVID-19 may indicate enterocyte functional change rather than cell death. Sci. Rep. 12:18792. 10.1038/s41598-022-23282-x - DOI - PMC - PubMed
-
- Asuni, N., and Wilder S.. 2019. VariantKey: A reversible numerical representation of human genetic variants. bioRxiv. 10.1101/473744 (Preprint posted February 15, 2019). - DOI
MeSH terms
Substances
Supplementary concepts
Grants and funding
- MR/S032304/1/UK Research and Innovation
- Francis Crick Institute
- UL1 TR001866/TR/NCATS NIH HHS/United States
- 108439/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- FC001003/CRUK_/Cancer Research UK/United Kingdom
- Imagine Institute
- R01 AI163029/AI/NIAID NIH HHS/United States
- JPB Foundation
- Fisher Center for Alzheimer's Research Foundation
- 202131-32-33/Fundació La Marató de TV3
- EC-GA no. 279185/European Union's Seventh Framework Programme
- R33HD105590/National Institute for Child Heath and Development
- EQU201903007798/French Foundation for Medical Research
- PA0873/NIHR Imperial Biomedical Research Centre
- Rockefeller University
- EQU202103012670/Fondation pour la recherche Medicale
- Imperial College London
- 58READIE/MRC_/Medical Research Council/United Kingdom
- Meyer Foundation
- ES/M001660/1/Economic and Social Research Council
- Generalitat de Catalunya
- Battersea & Bowery Advisory Group
- R01AI163029/NH/NIH HHS/United States
- Grandir - Fonds de solidarité pour l'enfance
- 848196/European Union's Horizon 2020 research and innovation
- 2021SGR00899/Agencia de Gestión de Ayudas Universitarias y de Investigación
- Institut National de la Santé et de la Recherche Médicale
- R33 HD105590/HD/NICHD NIH HHS/United States
- R21 AI160576/AI/NIAID NIH HHS/United States
- French Ministry of Higher Education, Research, and Innovation
- HHMI/Howard Hughes Medical Institute/United States
- Square Foundation
- Fondation du Souffle
- SCOR Corporate Foundation for Science
- St. Giles Foundation
- MR/N01104X/1 2018-2020/Economic and Social Research Council
- General Atlantic Foundation
- 101057100/H2020 UNDINE HORIZON-HLTH-2021-DISEASE-04
- FC001003/WT_/Wellcome Trust/United Kingdom
- ACCI20-759/CIBERER
- ANR-10-IAHU-01/French National Research Agency
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
