

Molecular and genetic insights into human ovarian aging from single-nuclei multi-omics analyses
- PMID: 39578560
- PMCID: PMC11839473
- DOI: 10.1038/s43587-024-00762-5
Molecular and genetic insights into human ovarian aging from single-nuclei multi-omics analyses
Abstract
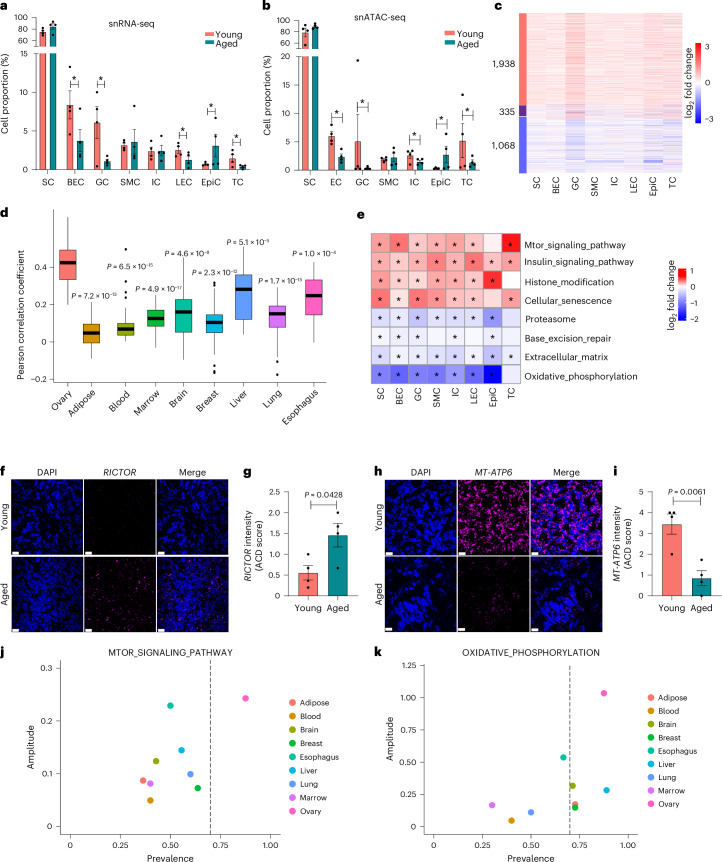
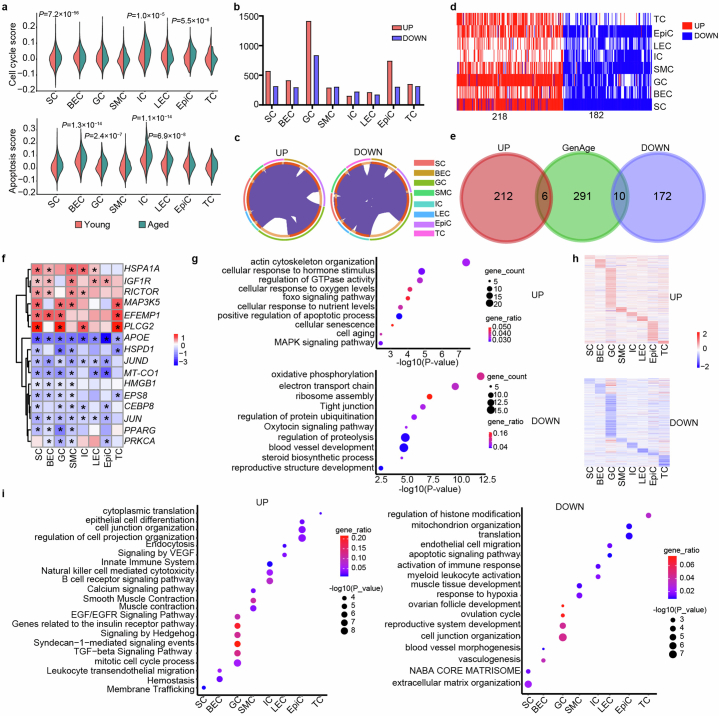
The ovary is the first organ to age in the human body, affecting both fertility and overall health. However, the biological mechanisms underlying human ovarian aging remain poorly understood. Here we present a comprehensive single-nuclei multi-omics atlas of four young (ages 23-29 years) and four reproductively aged (ages 49-54 years) human ovaries. Our analyses reveal coordinated changes in transcriptomes and chromatin accessibilities across cell types in the ovary during aging, notably mTOR signaling being a prominent ovary-specific aging pathway. Cell-type-specific regulatory networks reveal enhanced activity of the transcription factor CEBPD across cell types in the aged ovary. Integration of our multi-omics data with genetic variants associated with age at natural menopause demonstrates a global impact of functional variants on gene regulatory networks across ovarian cell types. We nominate functional non-coding regulatory variants, their target genes and ovarian cell types and regulatory mechanisms. This atlas provides a valuable resource for understanding the cellular, molecular and genetic basis of human ovarian aging.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures














References
-
- Spira, A. The decline of fecundity with age. MaturitasSuppl 1, 15–22 (1988). - PubMed
-
- Faddy, M., Gosden, R., Gougeon, A., Richardson, S. J. & Nelson, J. Accelerated disappearance of ovarian follicles in mid-life: implications for forecasting menopause. Hum. Reprod.7, 1342–1346 (1992). - PubMed
-
- Muka, T. et al. Association of age at onset of menopause and time since onset of menopause with cardiovascular outcomes, intermediate vascular traits, and all-cause mortality: a systematic review and meta-analysis. JAMA Cardiol.1, 767–776 (2016). - PubMed
MeSH terms
Substances
Grants and funding
- R01 AG057909/AG/NIA NIH HHS/United States
- P01 AG047200/AG/NIA NIH HHS/United States
- R01 AG057706/AG/NIA NIH HHS/United States
- R01 AG057433/AG/NIA NIH HHS/United States
- HL150521/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- DK127778/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 AG069750/AG/NIA NIH HHS/United States
- P01 AG017242/AG/NIA NIH HHS/United States
- AG057909/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 AG055501/AG/NIA NIH HHS/United States
- AG057341/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- AG17242/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- AG061521/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- RF1 AG057341/AG/NIA NIH HHS/United States
- R01 DK127778/DK/NIDDK NIH HHS/United States
- R01 HL150521/HL/NHLBI NIH HHS/United States
- AG057433/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- AG057706/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- AG055501/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 AG061521/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
