

Whole genome sequencing-based characterization and determination of quinolone resistance among methicillin-resistant and methicillin-susceptible S. Aureus isolates from patients attending regional referral hospitals in Tanzania
- PMID: 39578734
- PMCID: PMC11583670
- DOI: 10.1186/s12864-024-11045-z
Whole genome sequencing-based characterization and determination of quinolone resistance among methicillin-resistant and methicillin-susceptible S. Aureus isolates from patients attending regional referral hospitals in Tanzania
Abstract
Background: The emergence of multidrug-resistant termed Methicillin-resistant Staphylococcus aureus (MRSA) strain, driven by the acquisition of resistance gene mecA imposes a substantial challenge in the treatment and control of their related infections. Although quinolones have historically been effective against both MRSA and methicillin-susceptible S. aureus (MSSA) strains, the rising resistance to quinolones among S. aureus isolates, particularly in MRSA, has severely curtailed their potency and further narrowed down the therapeutic options. This study aimed to determine the burden of MRSA among isolates, as well as their resistance profile, genotypic characterization, and molecular relatedness through the construction of a phylogenetic tree.
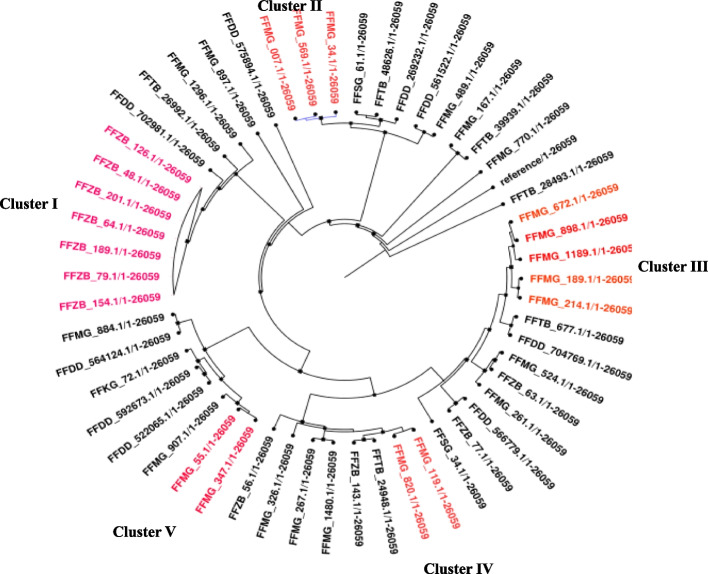
Materials and methods: Archived clinical S. aureus isolates from a descriptive, cross-sectional study involving six regional referral hospitals in Dodoma, Songea, Kigoma, Kitete, and Morogoro in the mainland Tanzania and Mnazi Mmoja in Zanzibar were analyzed. Bacterial identification was performed using both classical microbiology and whole genome sequencing on Illumina Nextseq 550 Sequencer. Species identification was done using KmerFinder 3.2, Multilocus Sequence Typing using MLST 2.0, SCCmec typing using SCCmecFinder 1.2, resistance genes using ResFinder 4.1, and phylogenetic relatedness using CSI Phylogeny 1.4.
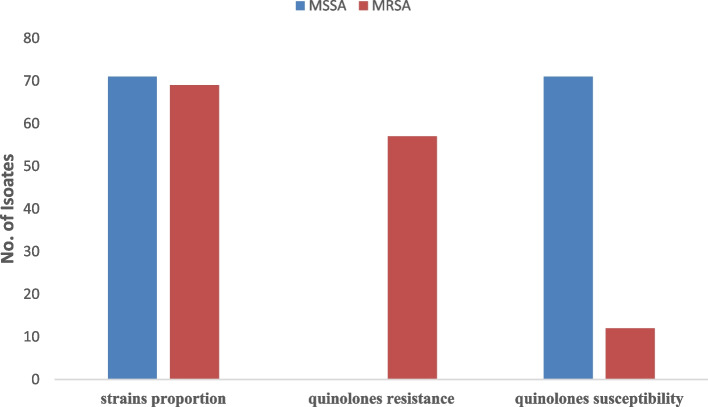
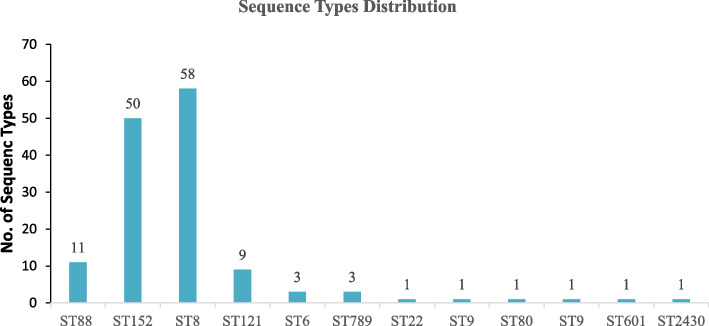
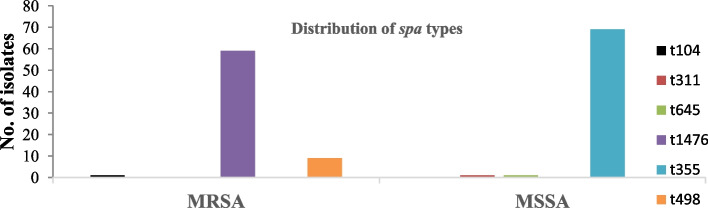
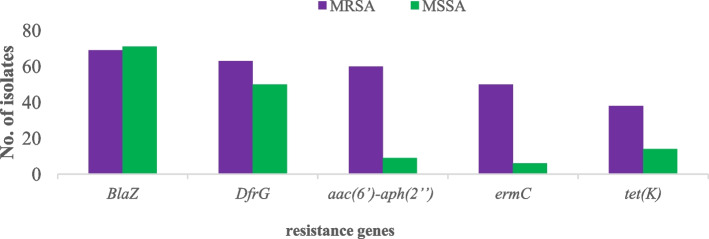
Results: Out of the 140 isolates analyzed, 69 (49.3%) were identified as MRSA, with 57 (82.6%) exhibiting quinolone resistance. Conversely, 71 isolates were identified as MSSA, and none of them exhibited resistance to quinolones. Spa-typing revealed six spa types, with t355, t1476, and t498 being the most common. Moreover, all (69) MRSA were found to carry SCCmec type IV. The isolates exhibited 14 different sequence types (STs). Notably, ST152 was prevalent among MSSA (50 isolates, 70%), while ST8 was the predominant sequence type among MRSA (58 isolates, 84%). The antimicrobial resistance profile revealed at least three horizontally acquired resistance genes, with blaZ, dfrG, tet(K), and aac (6')-aph (2'') genes being highly prevalent.
Conclusion: There is a high genetic diversity among the S. aureus isolates existing in Tanzania regional hospitals, with a concerning burden of quinolone resistance among MRSA isolates. The diversity in resistance genes among MRSA lineages emphasizes the necessity for the development of sustainable antimicrobial stewardship and surveillance to support evidence-based guidelines for managing and controlling MRSA infections in both community and hospital settings.
Keywords: Staphylococcus Cassette chromosome mec (SCCmec); Methicillin-resistant S. Aureus (MRSA); Sequence type (ST); Staphylococcal protein A (spa).
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The ethical clearance to conduct was sought from the Kilimanjaro Christian Medical University College (KCMUCo) Research Ethics and Review Committee and the ethical clearance with certificate number PG.171/2023 was given. Permission to access the Biotech laboratory and the archived samples was granted by the KCRI administration. The parent study secured its ethical clearance from NIMR Tanzania with reference number NIMR/HQ/R.8a/Vol.IX/3273. Informed consent was obtained from all participants or their legal guardians before the use of their samples for research purposes. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
