

Template-based modeling of insect odorant receptors outperforms AlphaFold3 for ligand binding predictions
- PMID: 39580516
- PMCID: PMC11585542
- DOI: 10.1038/s41598-024-80094-x
Template-based modeling of insect odorant receptors outperforms AlphaFold3 for ligand binding predictions
Abstract
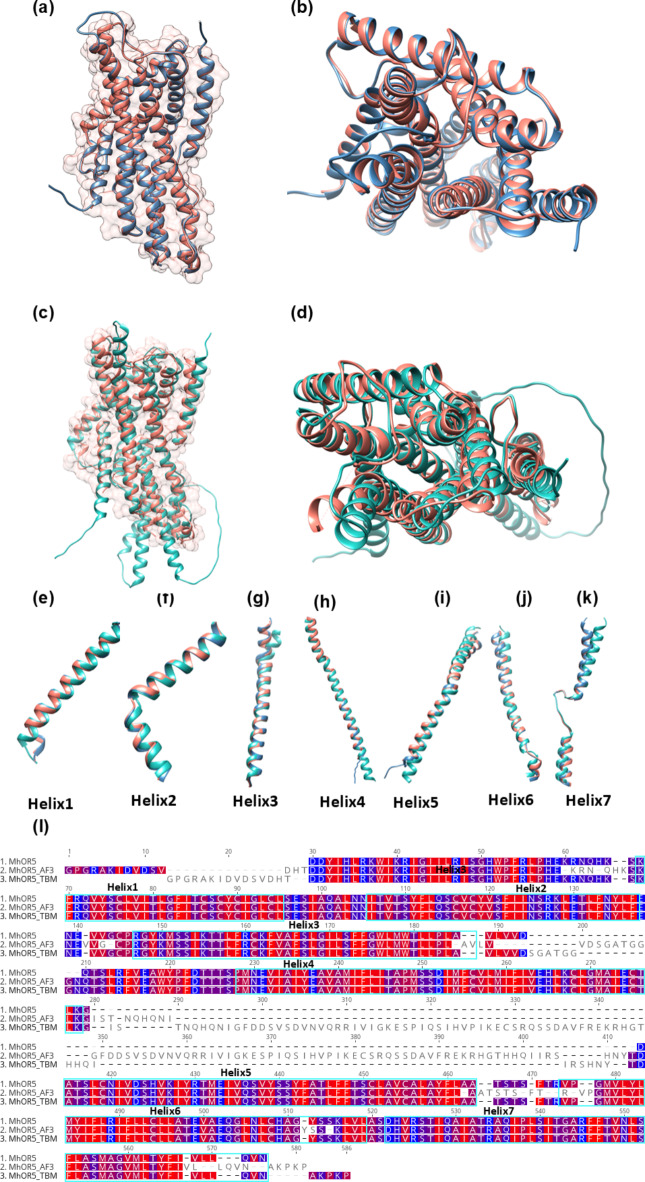
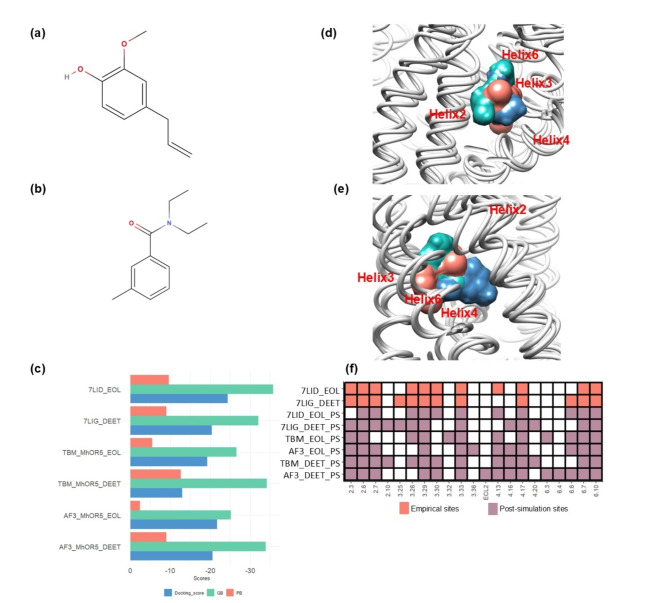
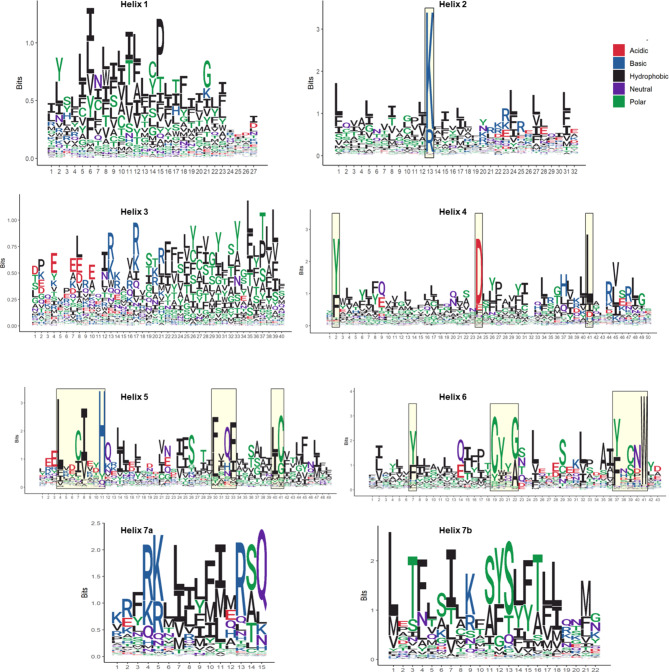
Insects rely on odorant receptors (ORs) to detect and respond to volatile environmental cues, so the ORs are attracting increasing interest as potential targets for pest control. However, experimental analysis of their structures and functions faces significant challenges. Computational methods such as template-based modeling (TBM) and AlphaFold3 (AF3) could facilitate the structural characterisation of ORs. This study first showed that both models accurately predicted the structural fold of MhOR5, a jumping bristletail OR with known experimental 3D structures, although accuracy was higher in the extracellular region of the protein and binding mode of their cognate ligands with TBM. The two approaches were then compared for their ability to predict the empirical binding evidence available for OR-odorant complexes in two economically important fruit fly species, Bactrocera dorsalis and B. minax. Post-simulation analyses including binding affinities, complex and ligand stability and receptor-ligand interactions (RLIs) revealed that TBM performed better than AF3 in discriminating between binder and non-binder complexes. TBM's superior performance is attributed to hydrophobicity-based helix-wise multiple sequence alignment (MSA) between available insect OR templates and the ORs for which the binding data were generated. This MSA identified conserved residues and motifs which could be used as anchor points for refining the alignments.
Keywords: Bactrocera; Molecular docking; Molecular dynamics; Receptor-ligand interactions; Transmembrane proteins.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures



Similar articles
-
Expression patterns and ligand binding characterization of Plus-C odorant-binding protein 14 from Adelphocoris lineolatus (Goeze).Comp Biochem Physiol B Biochem Mol Biol. 2019 Jan;227:75-82. doi: 10.1016/j.cbpb.2018.10.001. Epub 2018 Oct 5. Comp Biochem Physiol B Biochem Mol Biol. 2019. PMID: 30292754
-
Probing insect odorant receptors with their cognate ligands: insights into structural features.Biochem Biophys Res Commun. 2013 Jun 7;435(3):477-82. doi: 10.1016/j.bbrc.2013.05.015. Epub 2013 May 11. Biochem Biophys Res Commun. 2013. PMID: 23673297 Free PMC article.
-
The structural basis of odorant recognition in insect olfactory receptors.Nature. 2021 Sep;597(7874):126-131. doi: 10.1038/s41586-021-03794-8. Epub 2021 Aug 4. Nature. 2021. PMID: 34349260 Free PMC article.
-
Towards an understanding of the structural basis for insect olfaction by odorant receptors.Insect Biochem Mol Biol. 2015 Nov;66:31-41. doi: 10.1016/j.ibmb.2015.09.010. Epub 2015 Sep 28. Insect Biochem Mol Biol. 2015. PMID: 26416146 Review.
-
Structural aspects of sexual attraction and chemical communication in insects.Trends Biochem Sci. 2004 May;29(5):257-64. doi: 10.1016/j.tibs.2004.03.003. Trends Biochem Sci. 2004. PMID: 15130562 Review.
Cited by
-
Accelerating Ligand Discovery for Insect Odorant Receptors.Int J Biol Sci. 2025 Feb 18;21(5):2101-2117. doi: 10.7150/ijbs.105648. eCollection 2025. Int J Biol Sci. 2025. PMID: 40083686 Free PMC article.
-
AlphaFold 3: an unprecedent opportunity for fundamental research and drug development.Precis Clin Med. 2025 Jul 1;8(3):pbaf015. doi: 10.1093/pcmedi/pbaf015. eCollection 2025 Sep. Precis Clin Med. 2025. PMID: 40799364 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
