

Multi-omic and single-cell profiling of chromothriptic medulloblastoma reveals genomic and transcriptomic consequences of genome instability
- PMID: 39580568
- PMCID: PMC11585558
- DOI: 10.1038/s41467-024-54547-w
Multi-omic and single-cell profiling of chromothriptic medulloblastoma reveals genomic and transcriptomic consequences of genome instability
Erratum in
-
Author Correction: Multi-omic and single-cell profiling of chromothriptic medulloblastoma reveals genomic and transcriptomic consequences of genome instability.Nat Commun. 2025 Jan 27;16(1):1085. doi: 10.1038/s41467-025-56164-7. Nat Commun. 2025. PMID: 39870666 Free PMC article. No abstract available.
Abstract
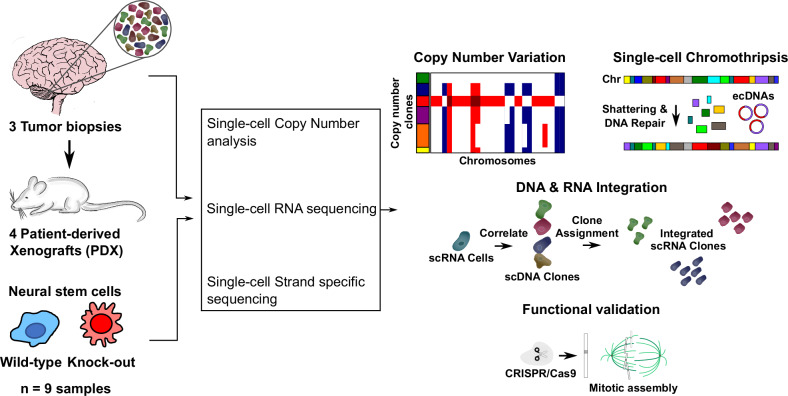
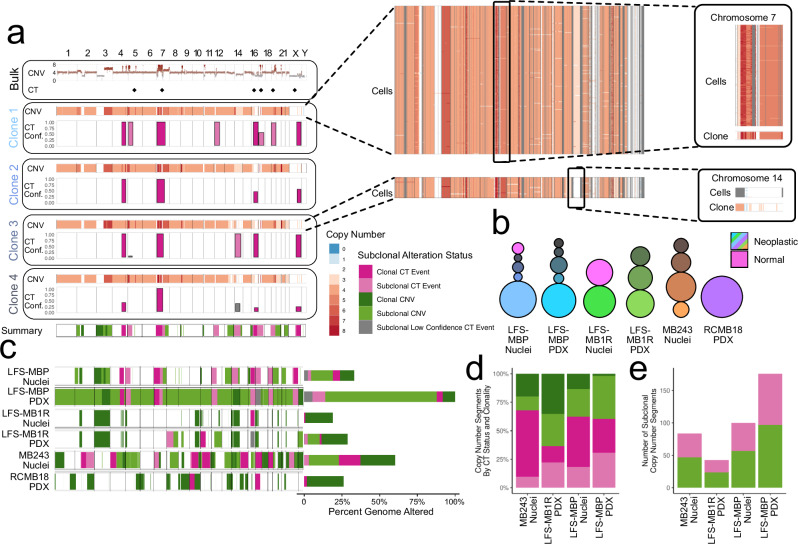
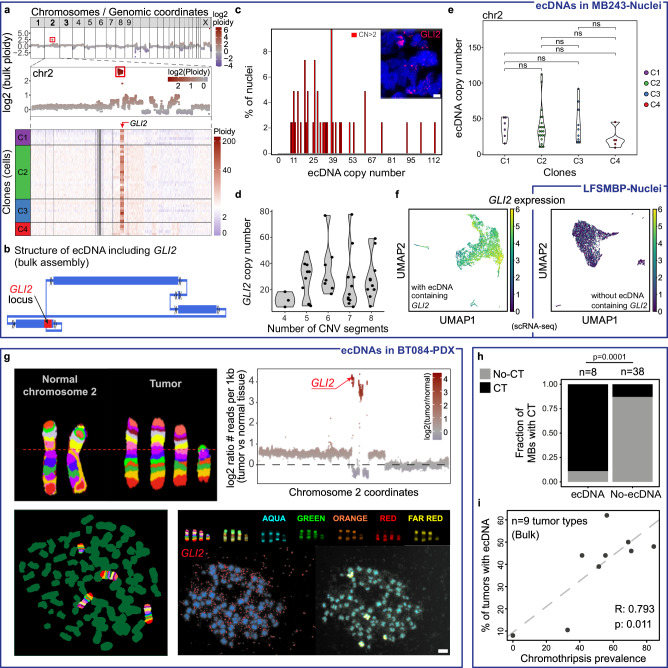
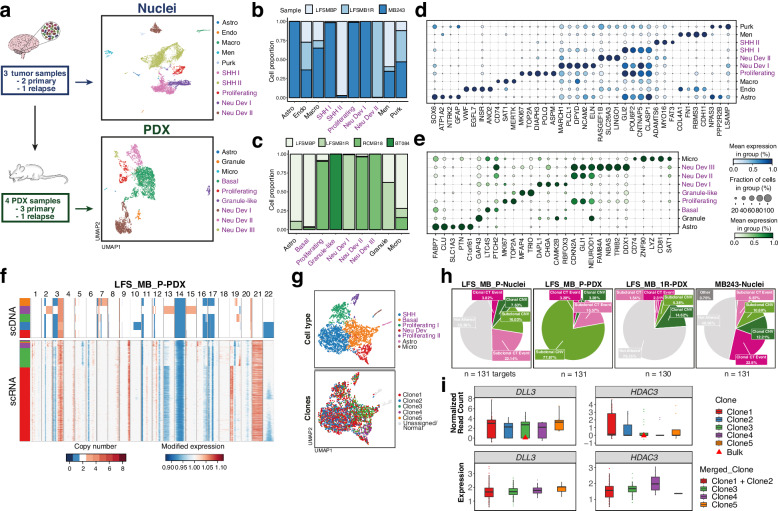
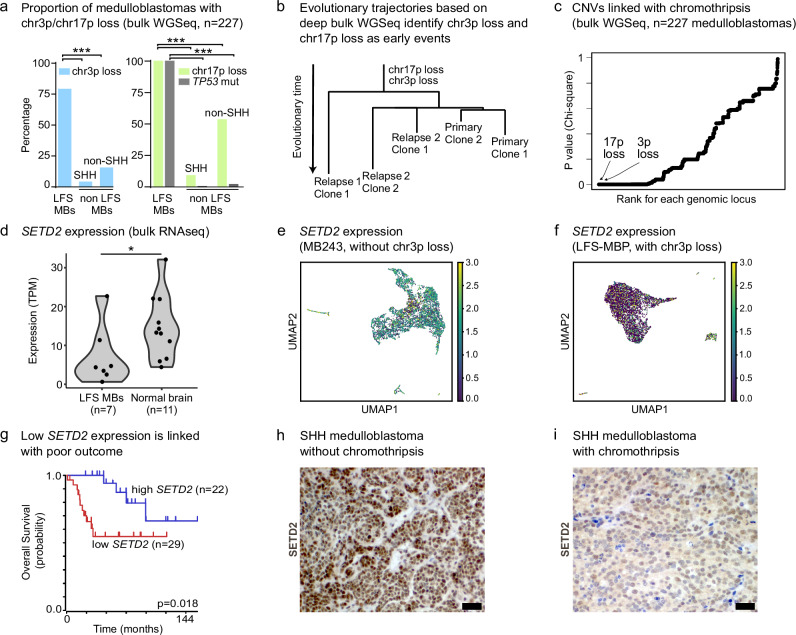
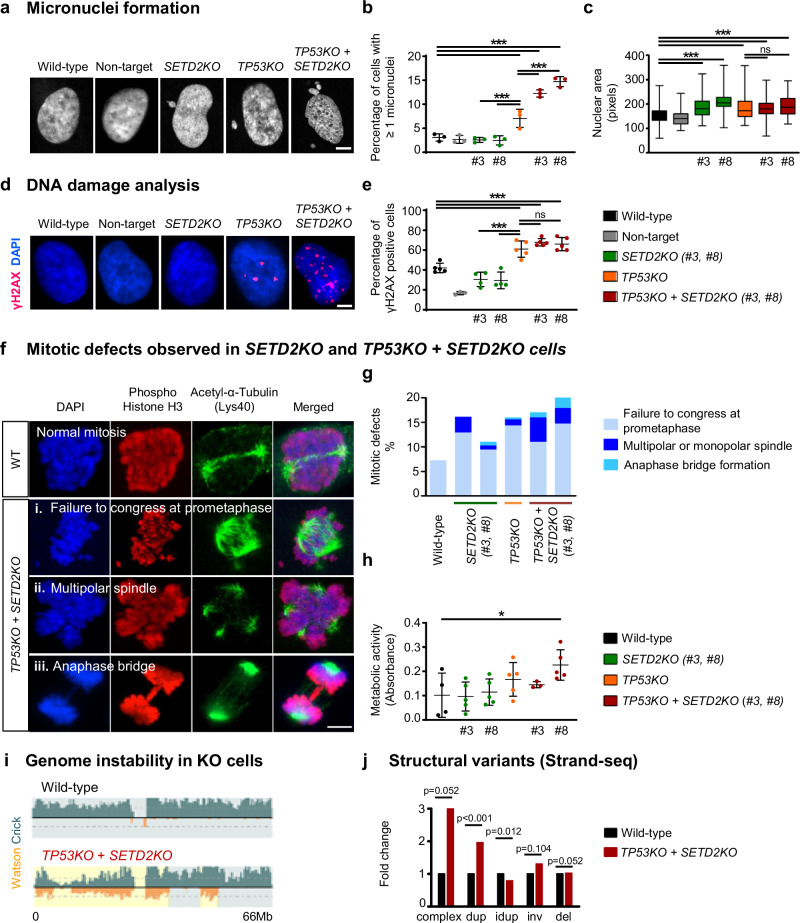
Chromothripsis is a frequent form of genome instability, whereby a presumably single catastrophic event generates extensive genomic rearrangements of one or multiple chromosome(s). However, little is known about the heterogeneity of chromothripsis across different clones from the same tumour, as well as changes in response to treatment. Here we analyse single-cell genomic and transcriptomic alterations linked with chromothripsis in human p53-deficient medulloblastoma and neural stem cells (n = 9). We reconstruct the order of somatic events, identify early alterations likely linked to chromothripsis and depict the contribution of chromothripsis to malignancy. We characterise subclonal variation of chromothripsis and its effects on extrachromosomal circular DNA, cancer drivers and putatively druggable targets. Furthermore, we highlight the causative role and the fitness consequences of specific rearrangements in neural progenitors.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
-
- Kloosterman, W. P., Koster, J. & Molenaar, J. J. Prevalence and clinical implications of chromothripsis in cancer genomes. Curr. Opin. Oncol.26, 64–72 (2014). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
