

Development of mutated β-catenin gene signature to identify CTNNB1 mutations from whole and spatial transcriptomic data in patients with HCC
- PMID: 39583094
- PMCID: PMC11582745
- DOI: 10.1016/j.jhepr.2024.101186
Development of mutated β-catenin gene signature to identify CTNNB1 mutations from whole and spatial transcriptomic data in patients with HCC
Abstract
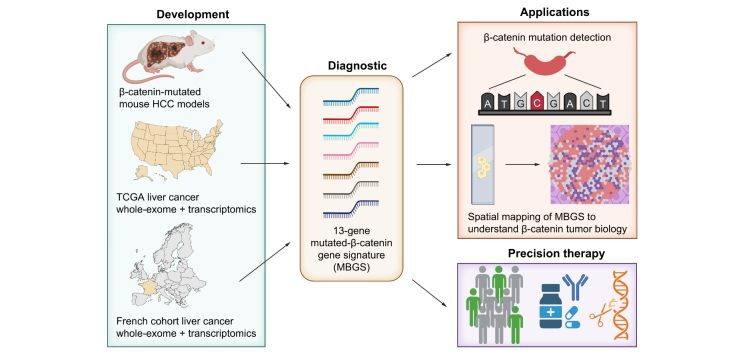
Background & aims: Patients with β-catenin (encoded by CTNNB1)-mutated hepatocellular carcinoma (HCC) demonstrate heterogenous responses to first-line immune checkpoint inhibitors (ICIs). Precision-medicine based treatments for this subclass are currently in clinical development. Here, we report derivation of the Mutated β-catenin Gene Signature (MBGS) to predict CTNNB1-mutational status in patients with HCC for future application in personalized medicine treatment regimens.
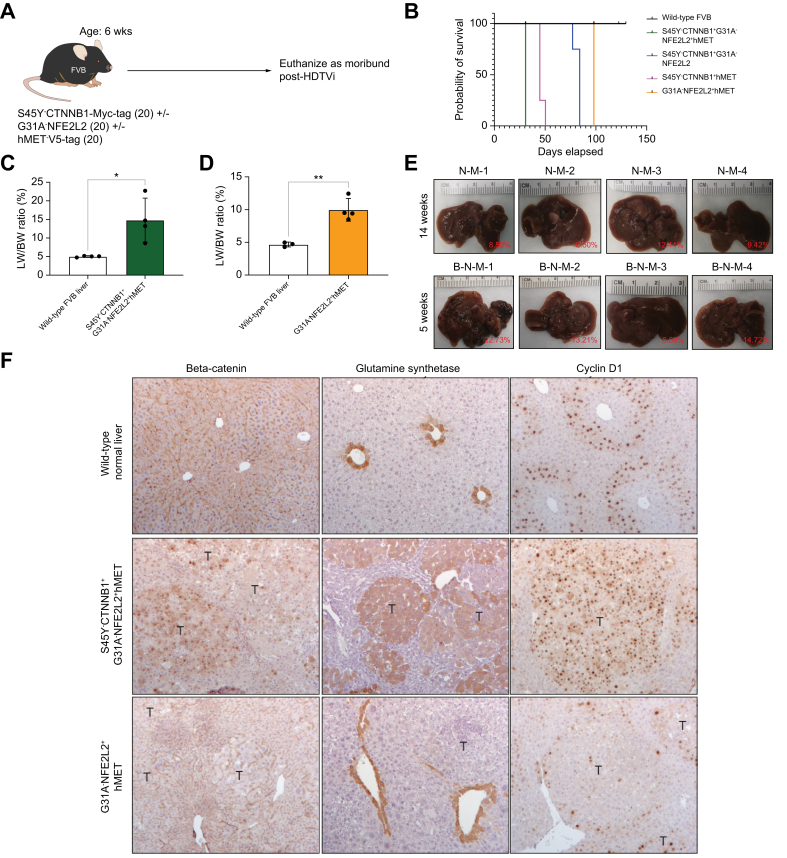
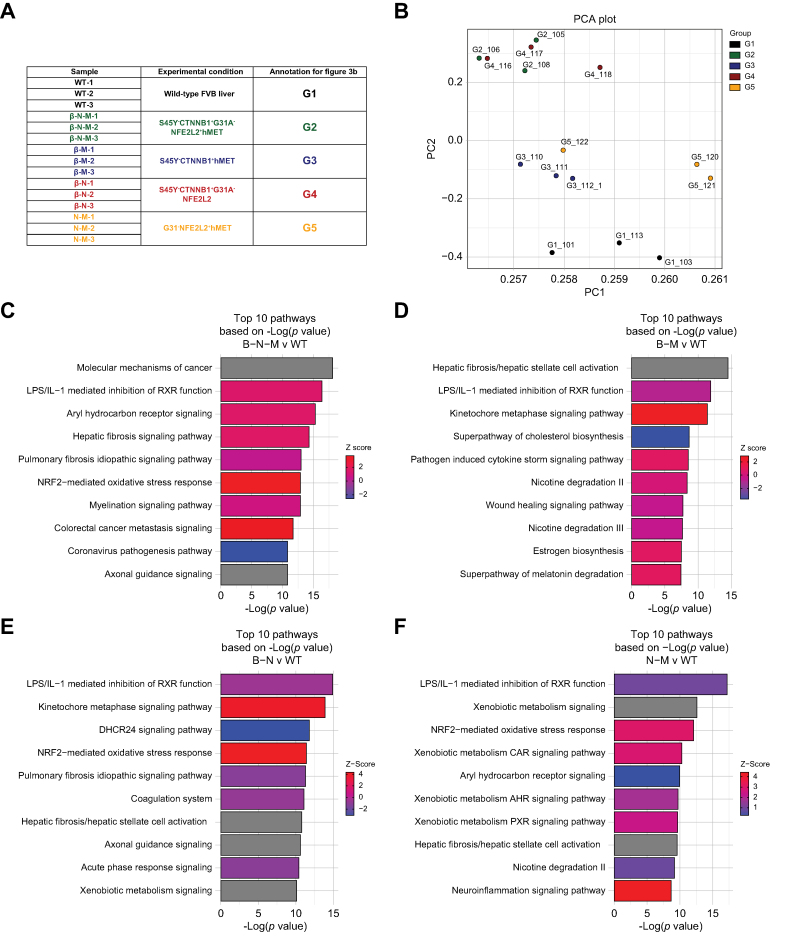
Methods: Co-expression of mutant-Nrf2 and hMet ± mutant-β-catenin in murine livers in mice led to HCC development. The MBGS was derived using bulk RNA-seq and intersectional transcriptomic analysis of β-catenin-mutated and non-mutated HCC models. Integrated RNA/whole-exome-sequencing and spatial transcriptomic data from multiple cohorts of patients with HCC was assessed to address the ability of MBGS to detect CTNNB1 mutation, the tumor immune microenvironment, and/or predict therapeutic responses.
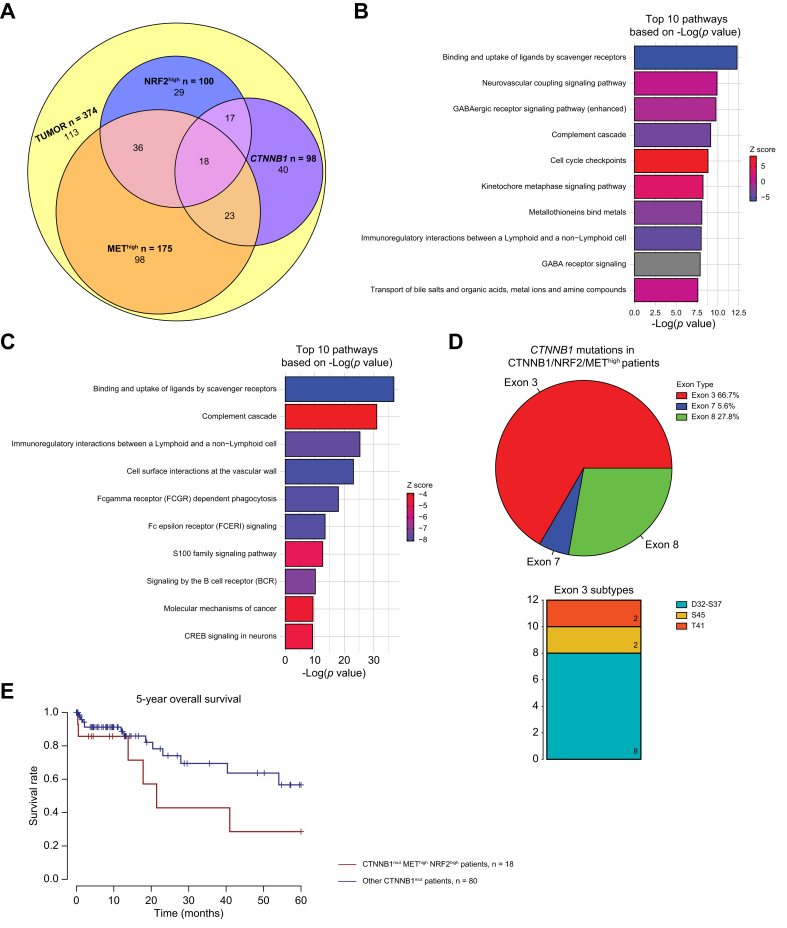
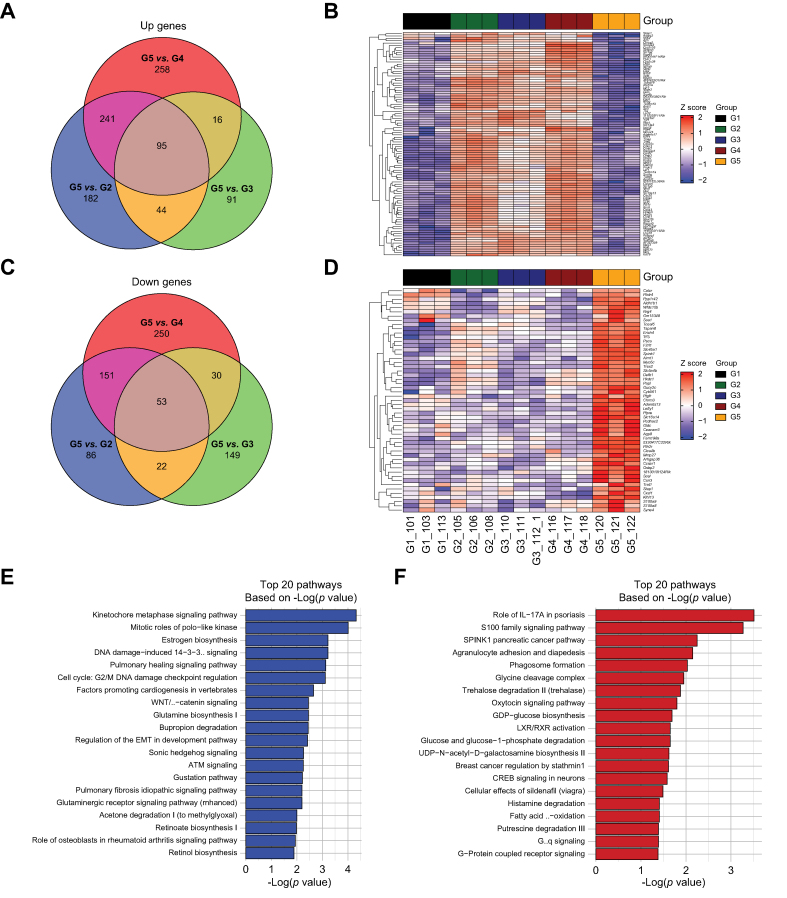
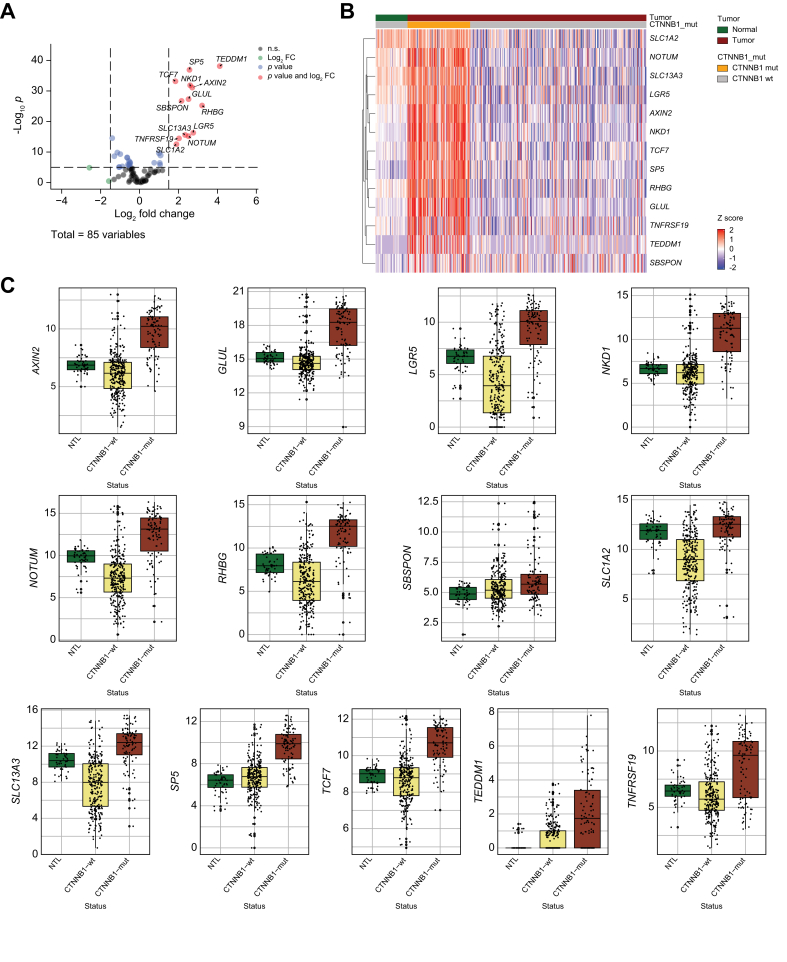
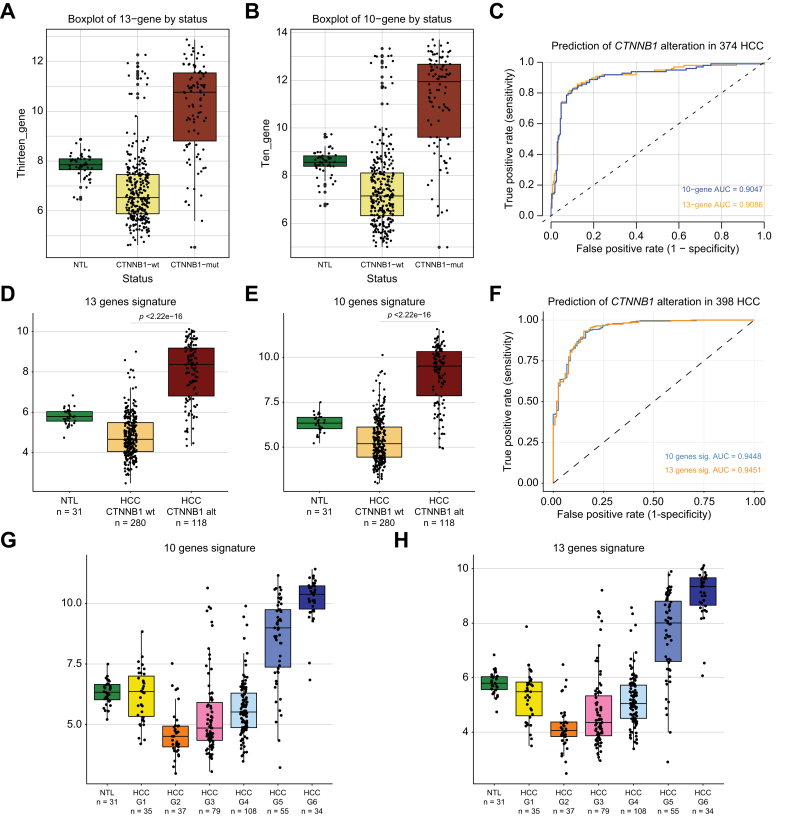
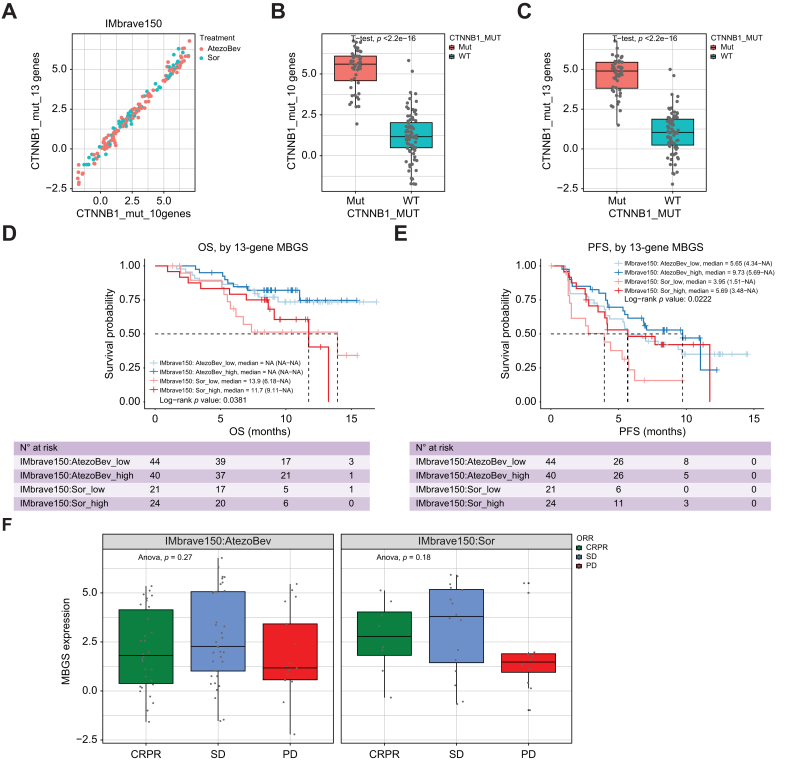
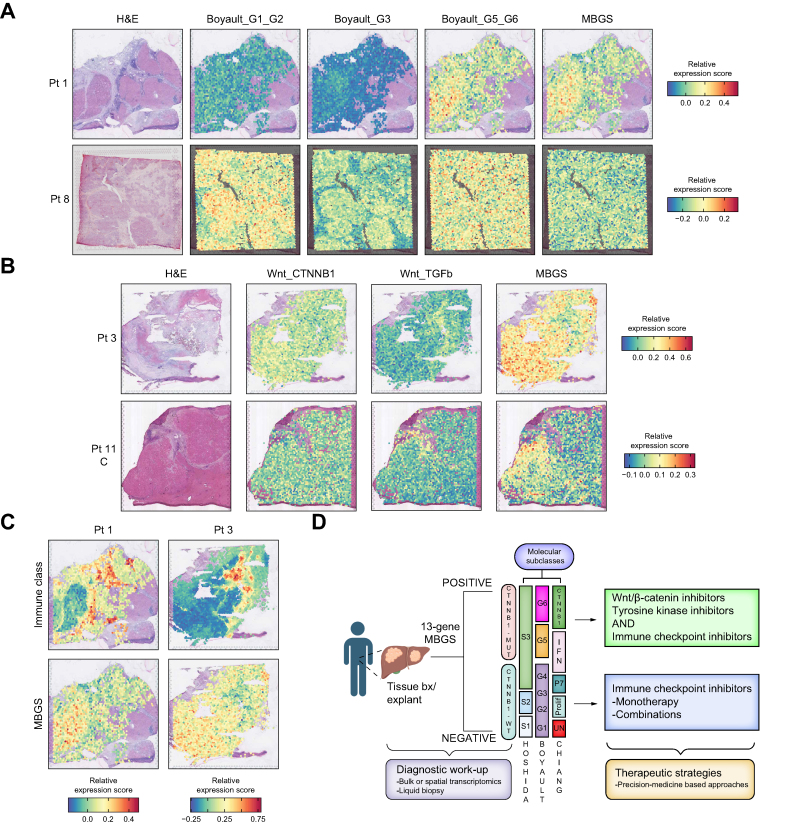
Results: Bulk RNA-seq comparing HCC specimens in mutant β-catenin-Nrf2, β-catenin-Met and β-catenin-Nrf2-Met to Nrf2-Met HCC model yielded 95 common upregulated genes. In The Cancer Genome Atlas (TCGA)-LIHC dataset, differential gene expression analysis with false discovery rate (FDR) = 0.05 and log2(fold change) >1.5 on the 95 common genes comparing CTNNB1-mutated vs. wild-type patients narrowed the gene panel to a 13-gene MBGS. MBGS predicted CTNNB1-mutations in TCGA (n = 374) and French (n = 398) patient cohorts with AUCs of 0.90 and 0.94, respectively. Additionally, a higher MBGS expression score was associated with lack of significant improvement in overall survival or progression-free survival in the atezolizumab-bevacizumab arm vs. the sorafenib arm in the IMbrave150 cohort. MBGS performed comparable or superior to other CTNNB1-mutant classifiers. MBGS overlapped with Hoshida S3, Boyault G5/G6, and Chiang CTNNB1 subclass tumors in TCGA and in HCC spatial transcriptomic datasets visually depicting these tumors to be situated in an immune excluded tumor microenvironment.
Conclusions: MBGS will aid in patient stratification to guide precision medicine therapeutics for CTNNB1-mutated HCC subclass as a companion diagnostic, as anti-β-catenin therapies become available.
Impact and implications: As precision medicine for liver cancer treatment becomes a reality, diagnostic tools are needed to help classify patients into groups for the best treatment choices. We have developed a molecular signature that could serve as a companion diagnostic and uses bulk or spatial transcriptomic data to identify a unique subclass of liver tumors. This subgroup of liver cancer patients derive limited benefit from the current standard of care and are expected to benefit from specialized directed therapies that are on the horizon.
Keywords: Gene signature; Hepatocellular carcinoma; Immunotherapy; Liver cancer; Precision medicine; Spatial transcriptomics; β-catenin.
© 2024 The Author(s).
Figures
References
-
- Sung H., Ferlay J., Siegel R.L., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. - PubMed
-
- Toh M.R., Wong E.Y., Wong S.H., et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology. 2023;164:766–782. - PubMed
-
- Llovet J.M., Zucman-Rossi J., Pikarsky E., et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2 - PubMed
-
- Cheng A.L., Qin S., Ikeda M., et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862–873. - PubMed
-
- Abou-Alfa G.K., Lau G., Kudo M., et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022;1 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
