

Hypoxia is linked to acquired resistance to immune checkpoint inhibitors in lung cancer
- PMID: 39585348
- PMCID: PMC11602551
- DOI: 10.1084/jem.20231106
Hypoxia is linked to acquired resistance to immune checkpoint inhibitors in lung cancer
Abstract
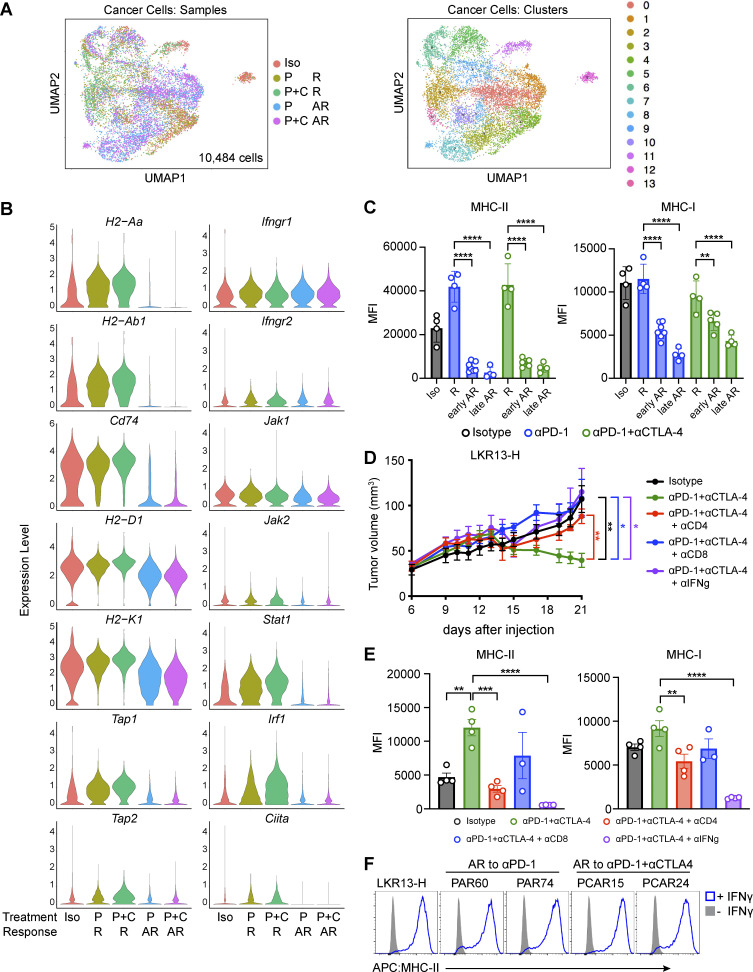
Despite the established use of immune checkpoint inhibitors (ICIs) to treat non-small cell lung cancer (NSCLC), only a subset of patients benefit from treatment and ∼50% of patients whose tumors respond eventually develop acquired resistance (AR). To identify novel drivers of AR, we generated murine Msh2 knock-out (KO) lung tumors that initially responded but eventually developed AR to anti-PD-1, alone or in combination with anti-CTLA-4. Resistant tumors harbored decreased infiltrating T cells and reduced cancer cell-intrinsic MHC-I and MHC-II levels, yet remained responsive to IFNγ. Resistant tumors contained extensive regions of hypoxia, and a hypoxia signature derived from single-cell transcriptional profiling of resistant cancer cells was associated with decreased progression-free survival in a cohort of NSCLC patients treated with anti-PD-1/PD-L1 therapy. Targeting hypoxic tumor regions using a hypoxia-activated pro-drug delayed AR to ICIs in murine Msh2 KO tumors. Thus, this work provides a rationale for targeting tumor metabolic features, such as hypoxia, in combination with immune checkpoint inhibition.
© 2024 Robles-Oteíza et al.
Conflict of interest statement
Disclosures: A. Ravi reported “other” from Halo Solutions outside the submitted work. F. Expósito reported being supported by the Fundación Ramón Areces Life and Matter Sciences Postdoctoral Fellowship and the 2023 LCRF leading edge Research Grant Program. N.D. Socci reported grants from NIH during the conduct of the study. T. Merghoub reported being a consultant for Immunos Therapeutics, Daiichi Sankyo Co, TigaTX, Normunity, and Pfizer. T. Merghoub is a cofounder of and equity holder in Imvaq Therapeutics. T. Merghoub receives research grant funding from Bristol Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, and Aprea Therapeutics. T. Merghoub is an inventor on patent applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neo-antigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4. M. Awad reported personal fees from Genentech, Bristol-Myers Squibb, Merck, AstraZeneca, Lilly, Mirati, Gritstone, EMD Serono, Regeneron, Janssen, Affini-T, Novartis, Coherus, D3Bio, and AbbVie; and grants from Amgen, Lilly, Bristol-Myers Squibb, Genentech, and AstraZeneca outside the submitted work. G. Getz reported research funds from IBM, Pharmacyclics/Abbvie, Bayer, Genentech, Calico, and Ultima Genomics, and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, MSEye, and MinimuMM-seq. G. Getz is a founder and consultant, and holds privately held equity in Scorpion Therapeutics; he is also a founder of, and holds privately held equity in, PreDICTA Biosciences. He was also a consultant to Merck. J. Gainor reported grants from Stand Up To Cancer and grants from Mark Foundation during the conduct of the study; personal fees from Bristol-Myers Squibb, Merck, Genentech/Roche, AstraZeneca, Loxo/Lilly, Mariana Therapeutics, Pfizer, Mirati Therapeutics, iTeos, Novartis, Merus Pharmaceuticals, AI proteins, Novocure, Silverback Therapeutics, Beigene, Takeda, and Sanofi; and grants from Moderna outside the submitted work; and that an immediate family member is an employee with equity in Ironwood Pharmaceuticals. M.D. Hellmann reported “other” from AstraZeneca outside the submitted work; and is an employee and stock holder of AstraZeneca. S.M. Kaech reported personal fees from EvolveImmune Therapeutics, Simcha Therapeutics, Siren Biotechnology, and Arvinas; and grants from Arvinas and Takeda during the conduct of the study. K. Politi reported grants from AstraZeneca, Roche/Genentech, Boehringer Ingelheim, and D2G Oncology; and personal fees from AstraZeneca and Revelio Therapeutics, Inc outside the submitted work; in addition, K. Politi had a patent to related to EGFR T790M mutation testing with royalties paid “from MSKCC/MolecularMD”; and serves on the AACR Board of Directors and the Lung Cancer Research Foundation Board of Directors. K. Politi is also a co-founder of and consultant for Revelio Therapeutics, Inc. No other disclosures were reported.
Figures










References
-
- Akce, M., Farran B., Switchenko J.M., Rupji M., Kang S., Khalil L., Ruggieri-Joyce A., Olson B., Shaib W.L., Wu C., et al. 2023. Phase II trial of nivolumab and metformin in patients with treatment-refractory microsatellite stable metastatic colorectal cancer. J. Immunother. Cancer. 11:e007235. 10.1136/jitc-2023-007235 - DOI - PMC - PubMed
-
- Augustin, R.C., Huang Z., Ding F., Zhai S., McArdle J., Santisi A., Davis M., Sander C., Davar D., Kirkwood J.M., et al. 2023. Metformin is associated with improved clinical outcomes in patients with melanoma: A retrospective, multi-institutional study. Front. Oncol. 13:1075823. 10.3389/fonc.2023.1075823 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- Stand Up To Cancer
- R01 CA195720/CA/NCI NIH HHS/United States
- F32 CA210516/CA/NCI NIH HHS/United States
- T32 CA009172/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA230275/CA/NCI NIH HHS/United States
- T32 CA193200/CA/NCI NIH HHS/United States
- K00 CA245819/CA/NCI NIH HHS/United States
- F99 CA245819/CA/NCI NIH HHS/United States
- T32 CA193200-01A1/National Institute of Health
- SU2C-AACR-DT17-15/American Cancer Society
- K99 CA245897/CA/NCI NIH HHS/United States
- 19-029-MIA/Mark Foundation For Cancer Research
- 2022R1A4A2000827/National Research Foundation of Korea
- American Association for Cancer Research
- R50 CA265359/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
