

Regulation of formin INF2 and its alteration in INF2-linked inherited disorders
- PMID: 39586895
- PMCID: PMC11589041
- DOI: 10.1007/s00018-024-05499-3
Regulation of formin INF2 and its alteration in INF2-linked inherited disorders
Abstract
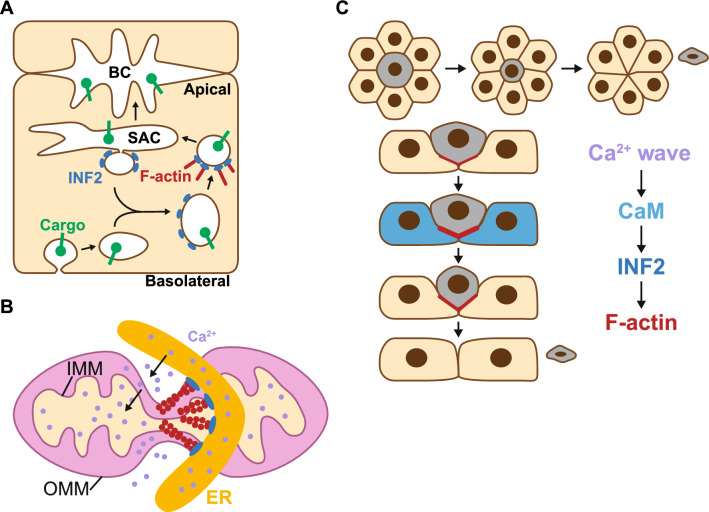
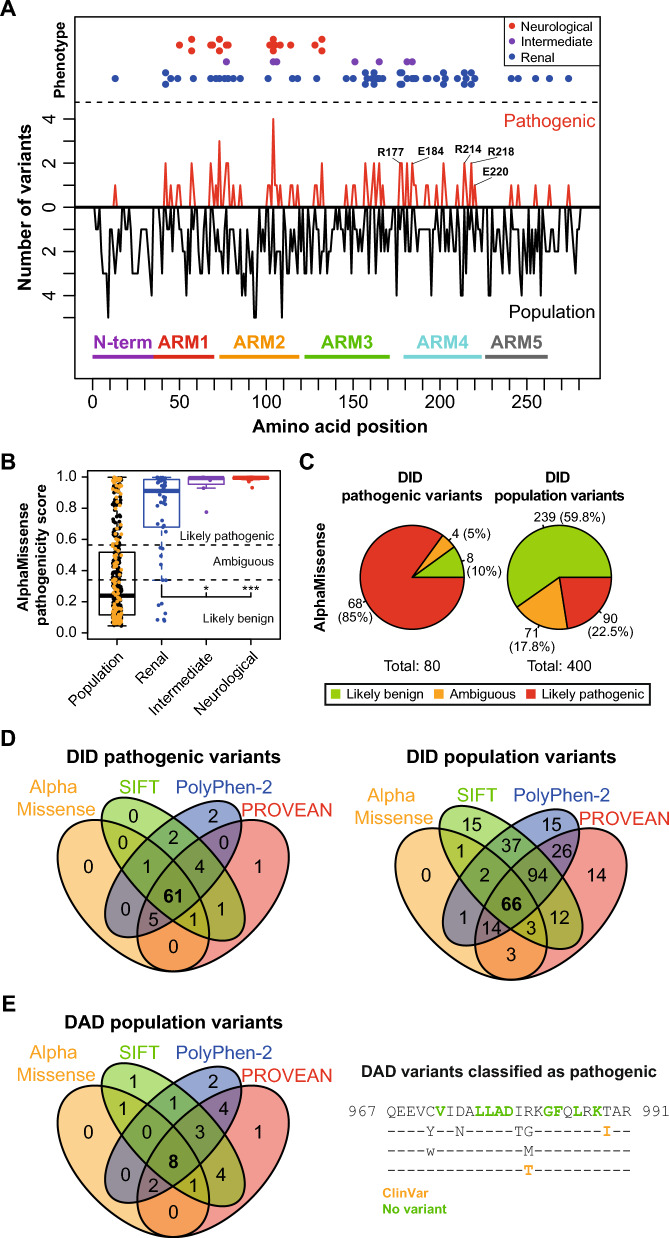
Formins are proteins that catalyze the formation of linear filaments made of actin. INF2, a formin, is crucial for correct vesicular transport, microtubule stability and mitochondrial division. Its activity is regulated by a complex of cyclase-associated protein and lysine-acetylated G-actin (KAc-actin), which helps INF2 adopt an inactive conformation through the association of its N-terminal diaphanous inhibitory domain (DID) with its C-terminal diaphanous autoinhibitory domain. INF2 activation can occur through calmodulin binding, KAc-actin deacetylation, G-actin binding, or association with the Cdc42 GTPase. Mutations in the INF2 DID are linked to focal segmental glomerulosclerosis (FSGS), affecting podocytes, and Charcot-Marie-Tooth disease, which affects Schwann cells and leads to axonal loss. At least 80 pathogenic DID variants of INF2 have been identified, with potential for many more. These mutations disrupt INF2 regulation, leading to excessive actin polymerization. This in turn causes altered intracellular trafficking, abnormal mitochondrial dynamics, and profound transcriptional reprogramming via the MRTF/SRF complex, resulting in mitotic abnormalities and p53-mediated cell death. This sequence of events could be responsible for progressive podocyte loss during glomerular degeneration in FSGS patients. Pharmacological targeting of INF2 or actin polymerization could offer the therapeutic potential to halt the progression of FSGS and improve outcomes for patients with INF2-linked disease.
Keywords: Actin; Charcot–Marie–Tooth disease; Focal segmental glomerulosclerosis; Mitotic catastrophe; Pathogenic variants; Podocytes.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Conflict of interest: The authors declare no competing interests. Ethical approval: Not applicable. Consent to participate: Not applicable. Consent for publication: Not applicable.
Figures
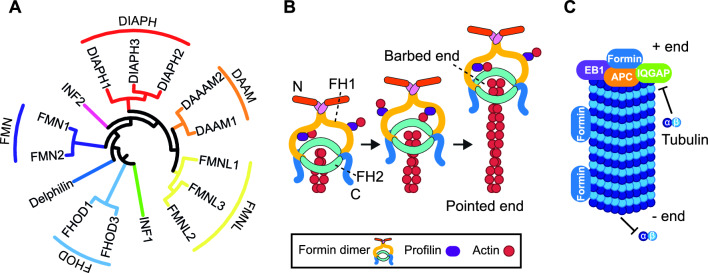
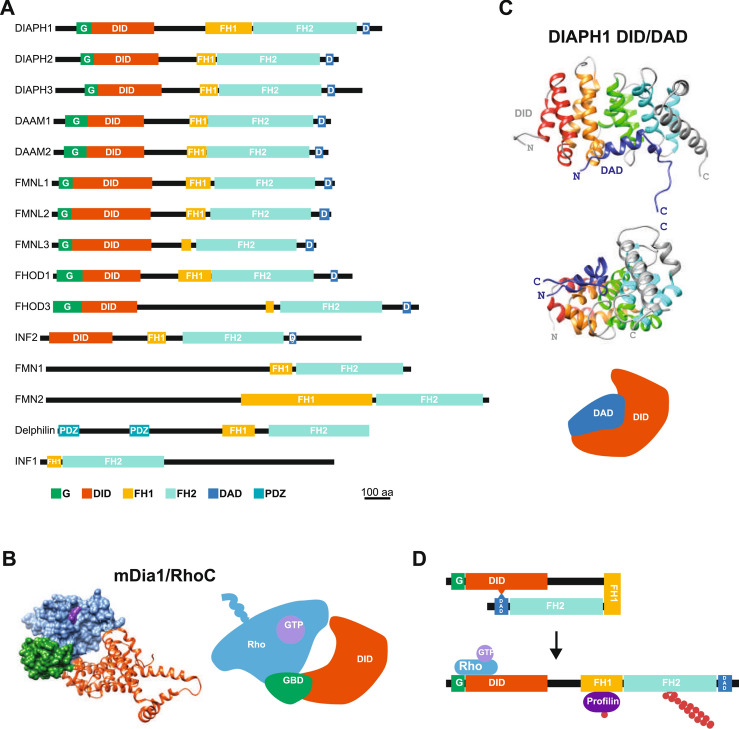
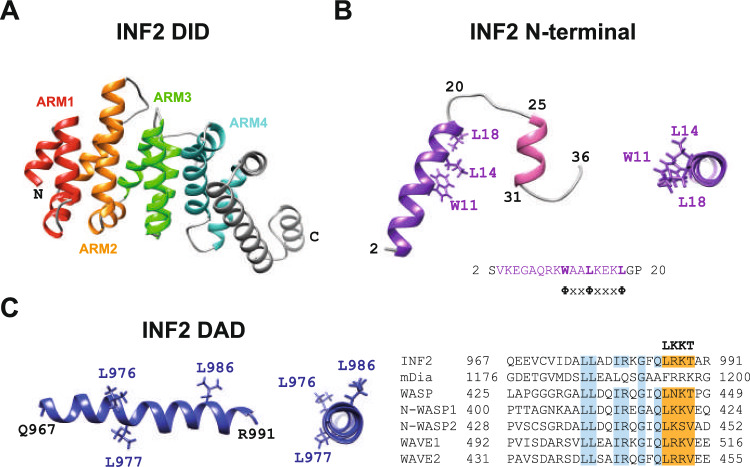

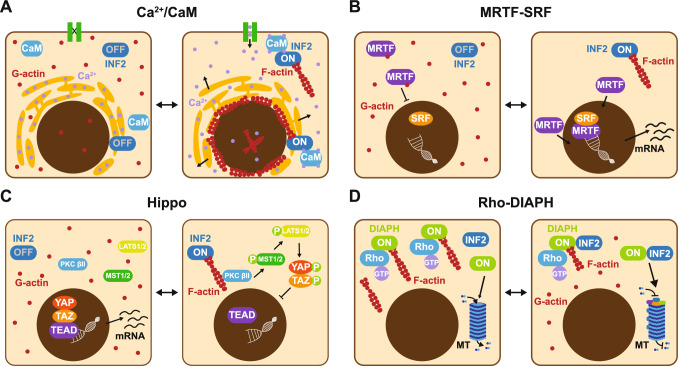

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
