

Single-cell multiomics analysis reveals dynamic clonal evolution and targetable phenotypes in acute myeloid leukemia with complex karyotype
- PMID: 39587361
- PMCID: PMC11631769
- DOI: 10.1038/s41588-024-01999-x
Single-cell multiomics analysis reveals dynamic clonal evolution and targetable phenotypes in acute myeloid leukemia with complex karyotype
Abstract
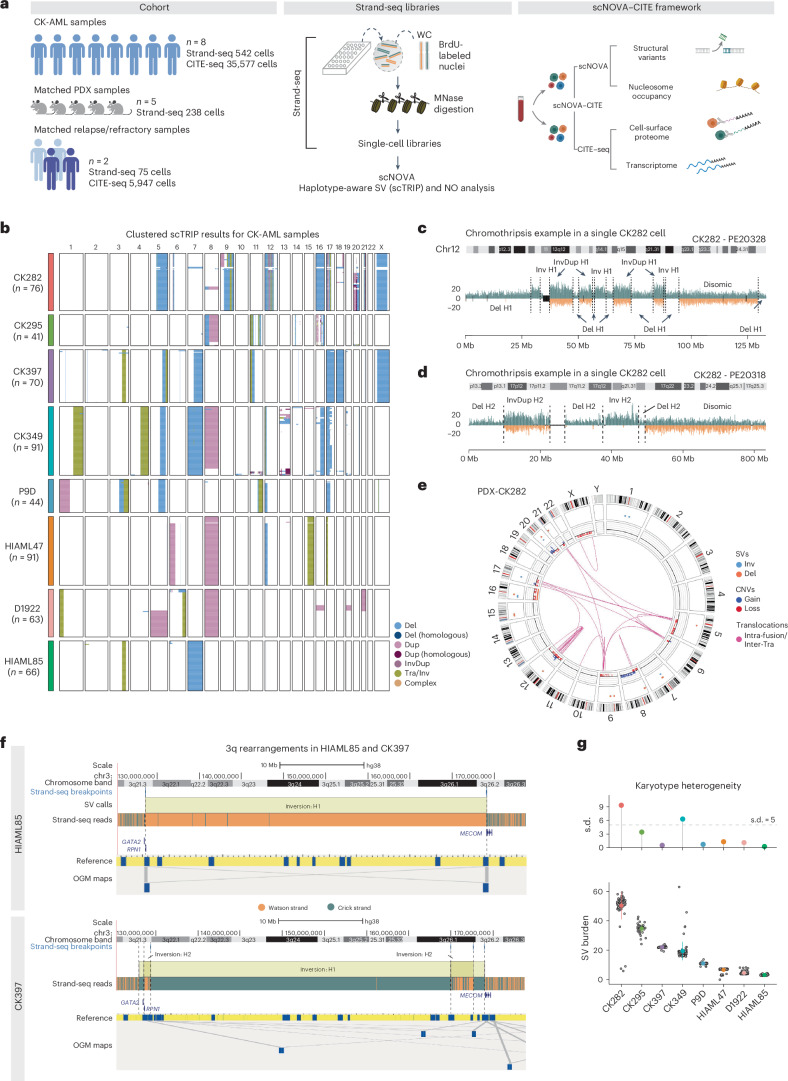
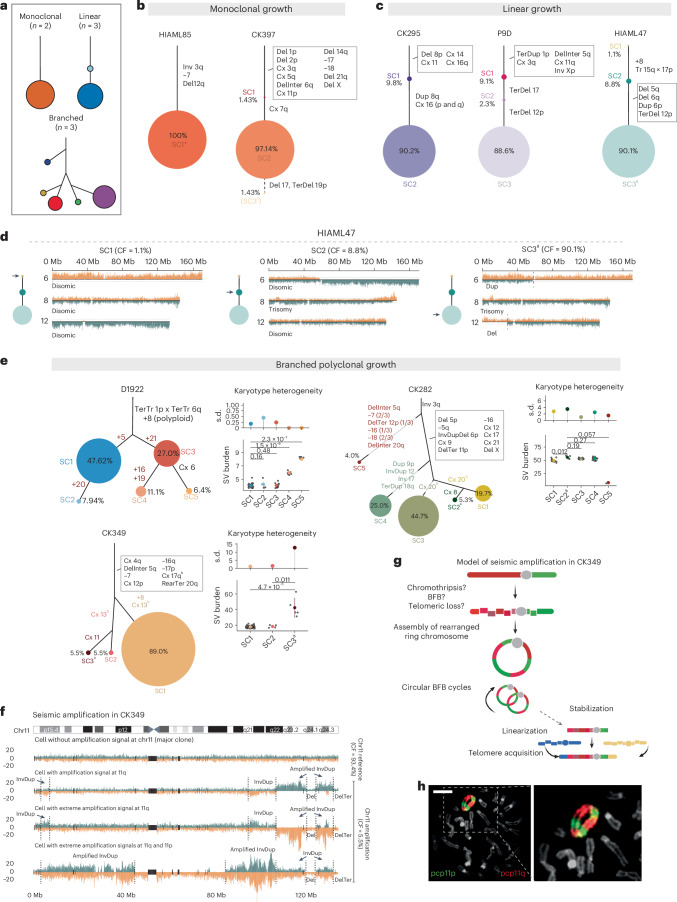
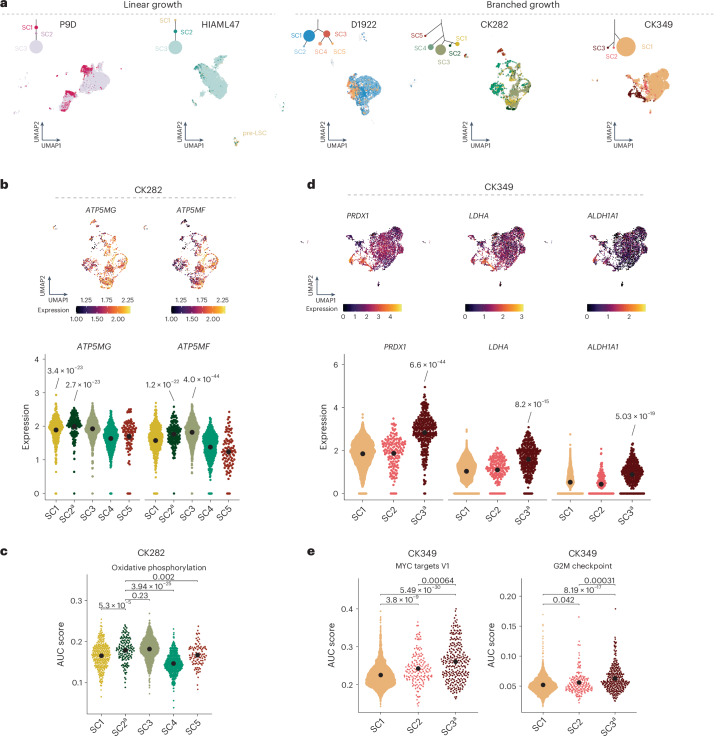
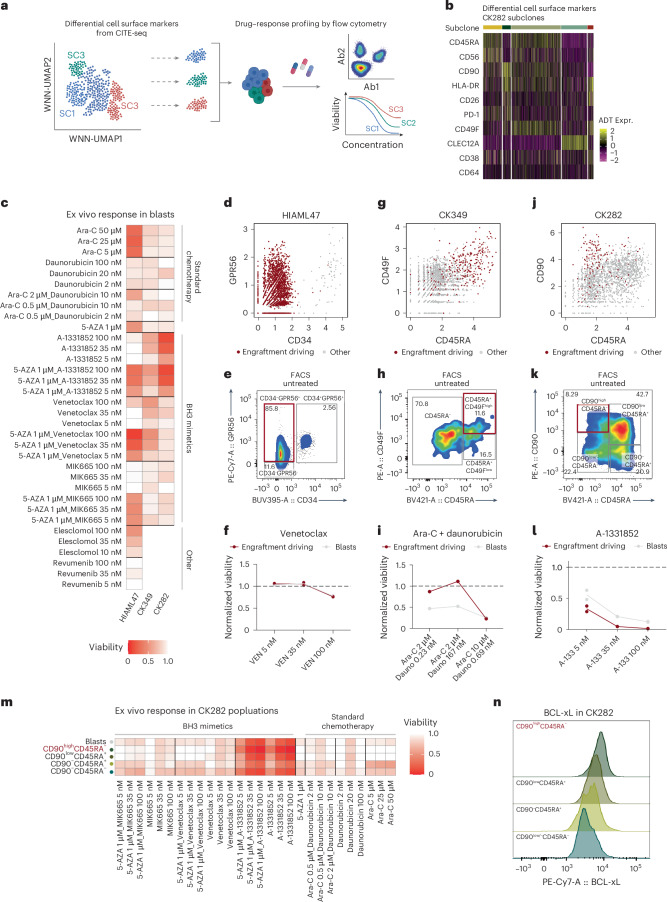
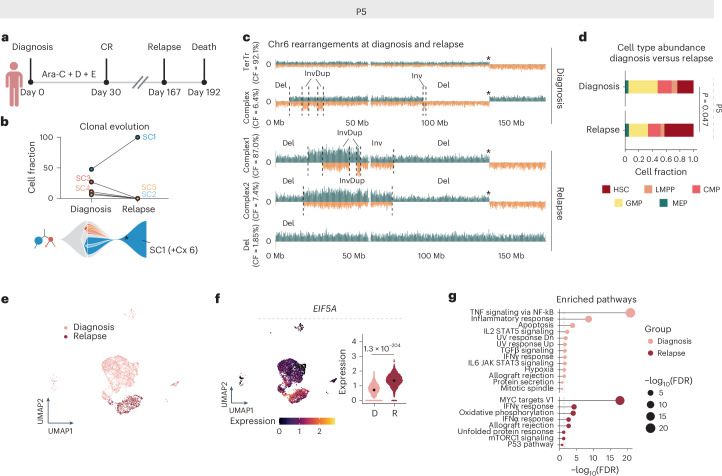
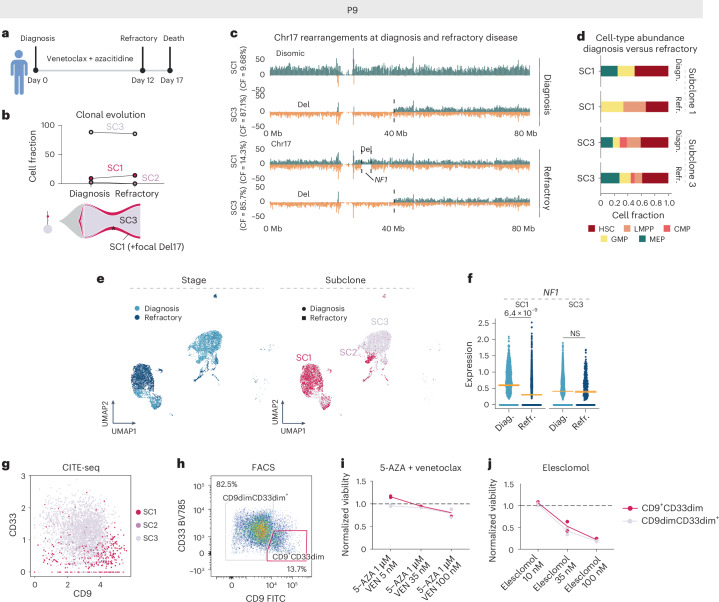
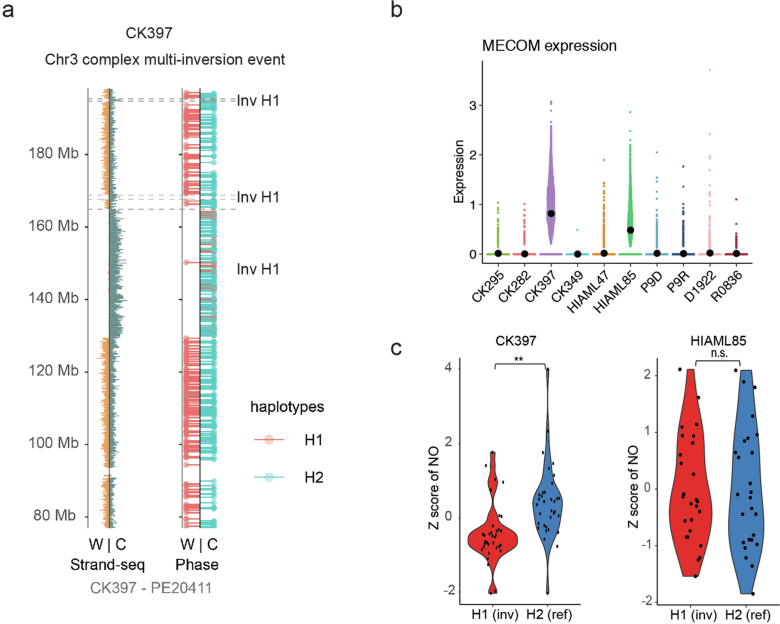
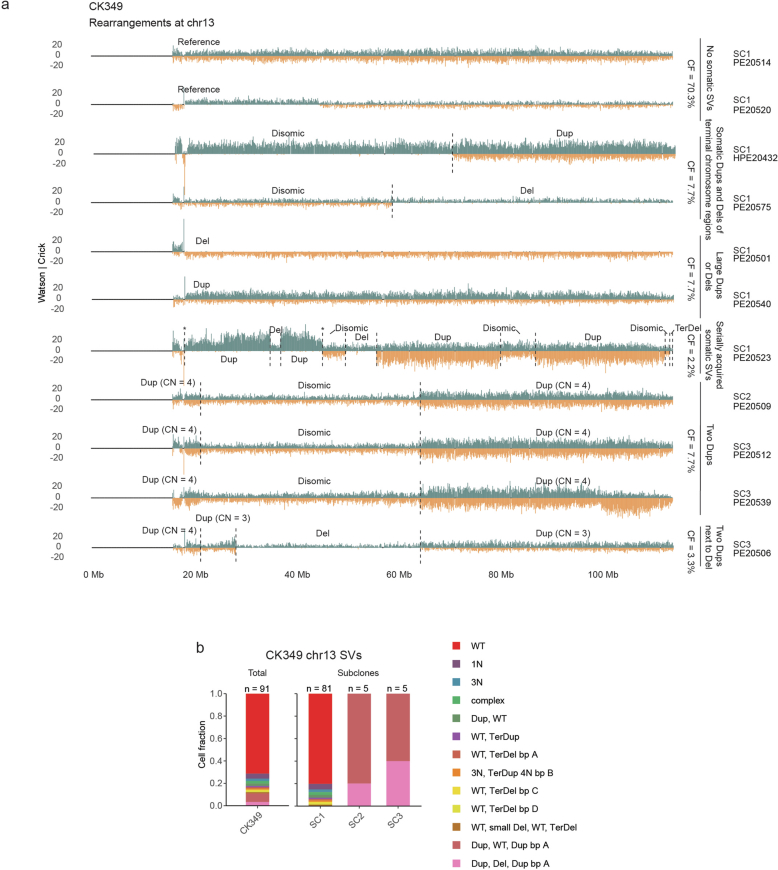
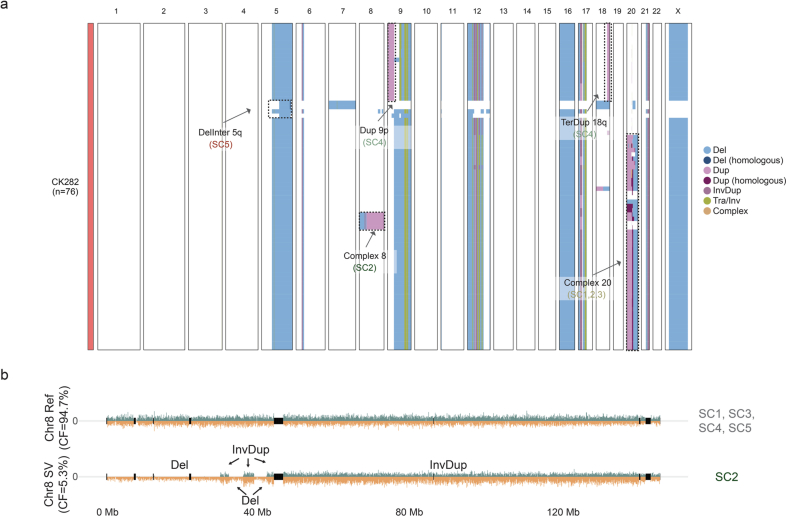
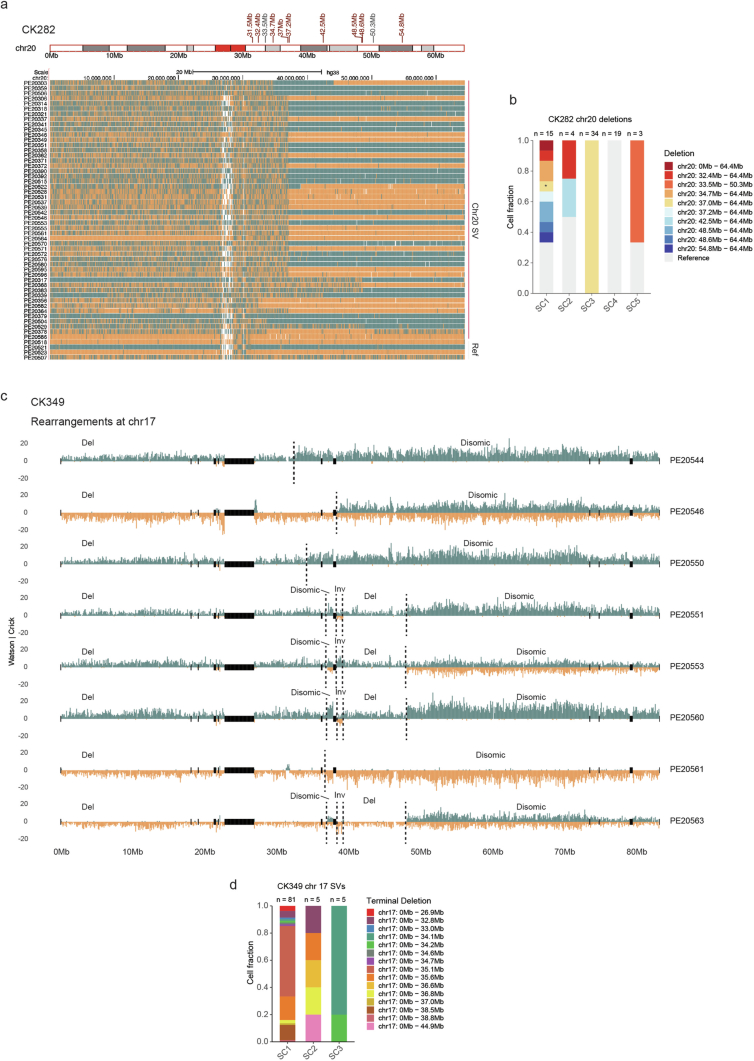
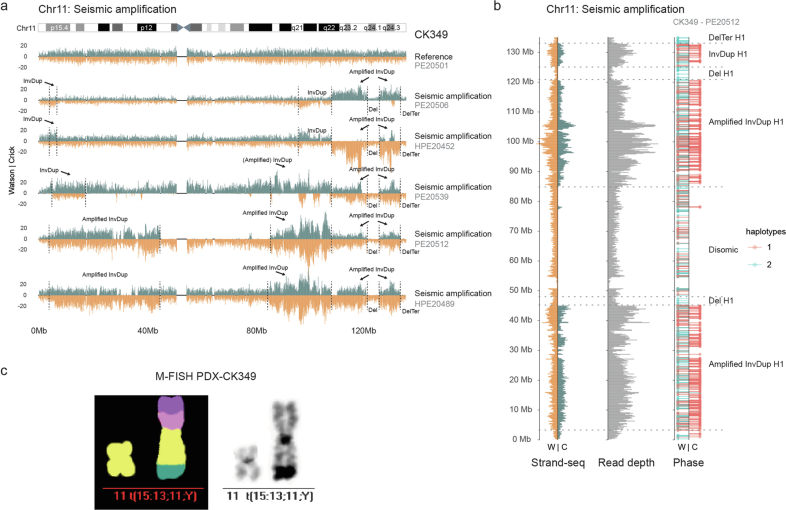
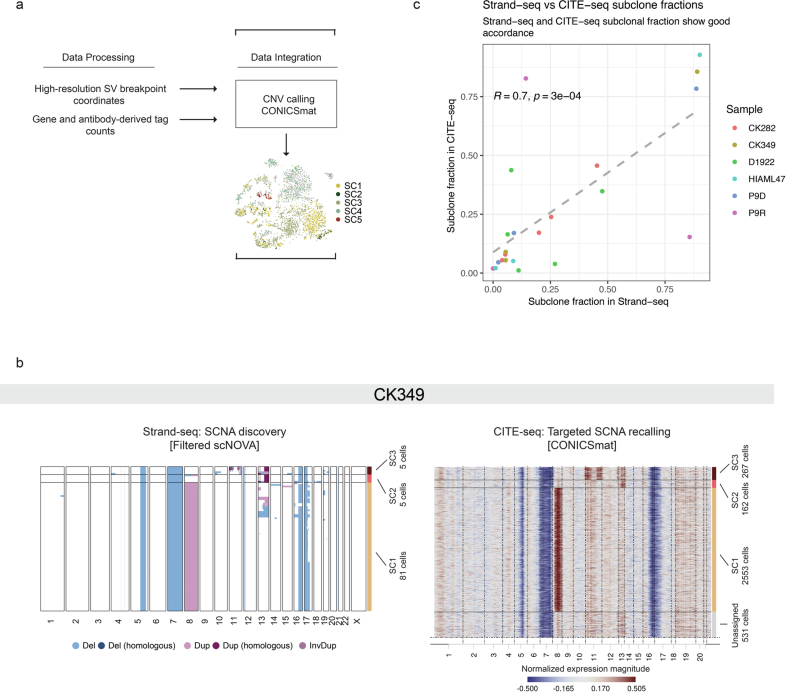
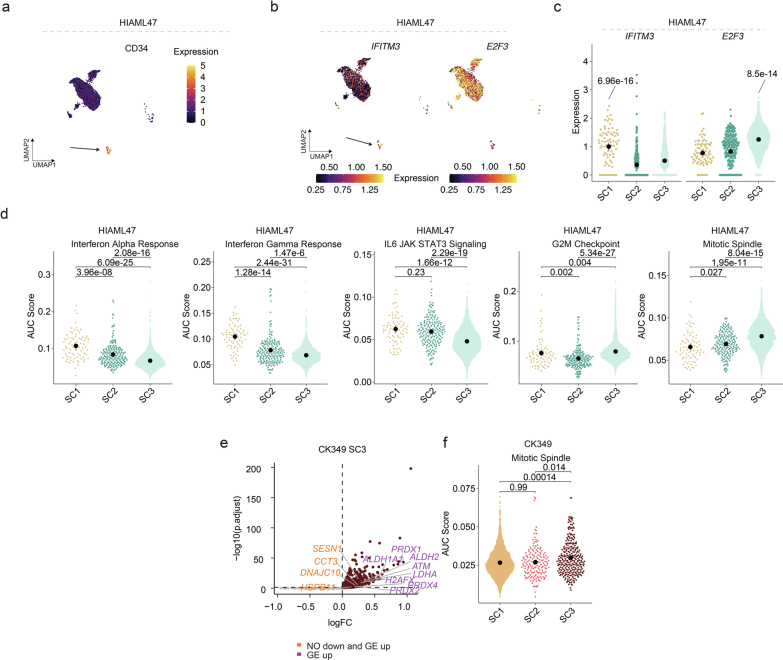
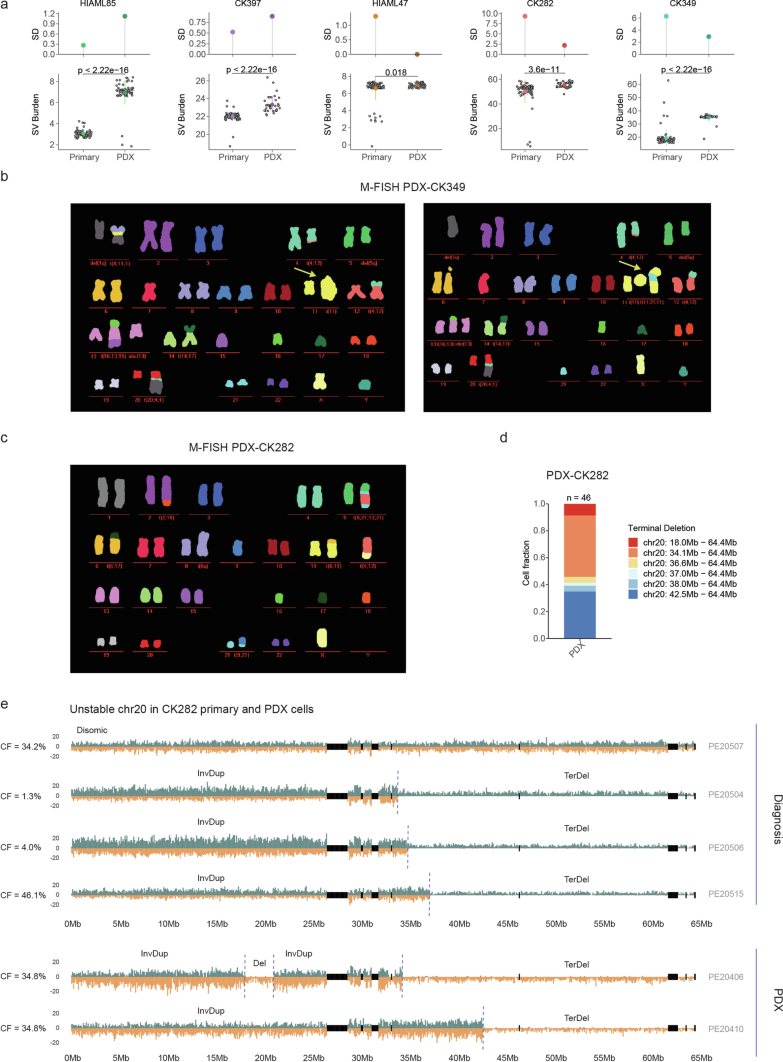
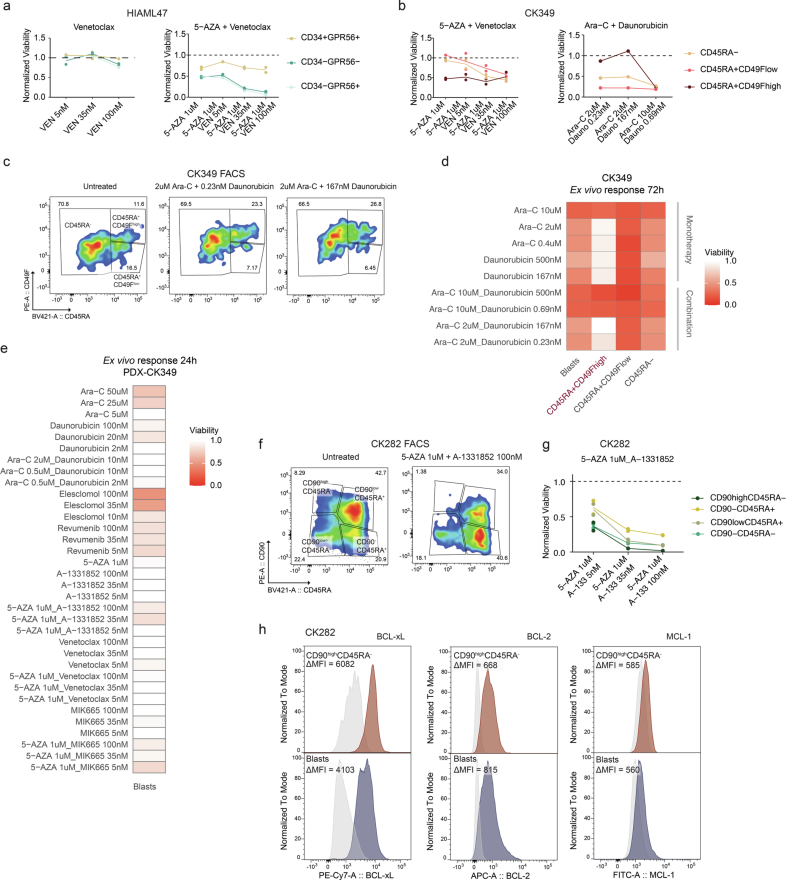
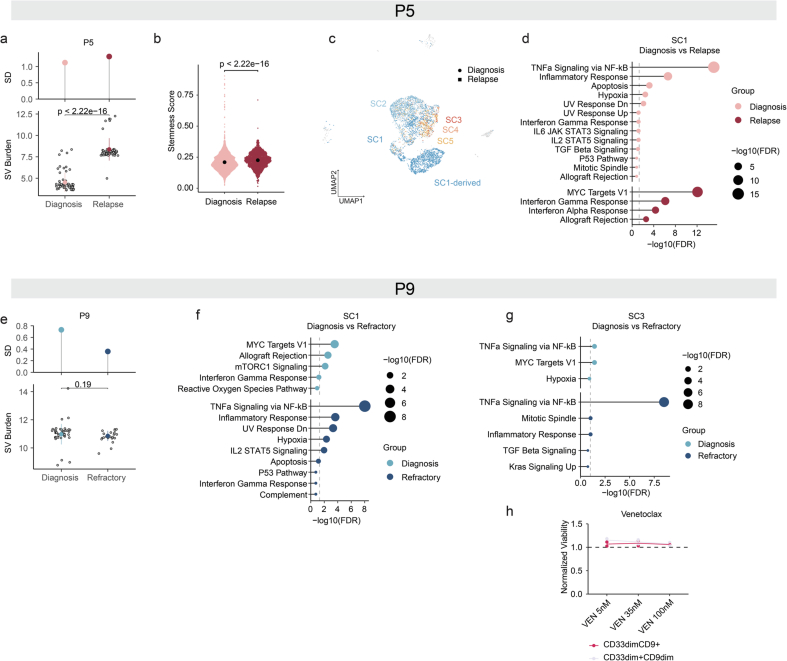
Chromosomal instability is a major driver of intratumoral heterogeneity (ITH), promoting tumor progression. In the present study, we combined structural variant discovery and nucleosome occupancy profiling with transcriptomic and immunophenotypic changes in single cells to study ITH in complex karyotype acute myeloid leukemia (CK-AML). We observed complex structural variant landscapes within individual cells of patients with CK-AML characterized by linear and circular breakage-fusion-bridge cycles and chromothripsis. We identified three clonal evolution patterns in diagnosis or salvage CK-AML (monoclonal, linear and branched polyclonal), with 75% harboring multiple subclones that frequently displayed ongoing karyotype remodeling. Using patient-derived xenografts, we demonstrated varied clonal evolution of leukemic stem cells (LSCs) and further dissected subclone-specific drug-response profiles to identify LSC-targeting therapies, including BCL-xL inhibition. In paired longitudinal patient samples, we further revealed genetic evolution and cell-type plasticity as mechanisms of disease progression. By dissecting dynamic genomic, phenotypic and functional complexity of CK-AML, our findings offer clinically relevant avenues for characterizing and targeting disease-driving LSCs.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: A.D.S. and J.O.K. have previously disclosed a patent application (no. EP19169090) that is relevant to this manuscript. A.K.E. received an honorarium from AstraZeneca for serving on their diversity, equity and inclusion advisory board, and her spouse has ownership interest and is employed by Karyopharm Therapeutics. The remaining authors declare no competing interests.
Figures

References
-
- Bochtler, T. et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J. Clin. Oncol.31, 3898–3905 (2013). - PubMed
-
- Rücker, F. G. et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood119, 2114–2121 (2012). - PubMed
MeSH terms
Grants and funding
- R01 CA284595/CA/NCI NIH HHS/United States
- R01 CA283574/CA/NCI NIH HHS/United States
- R01CA262496, R01CA284595-01, R01CA283574-01/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- SPP2036, FOR2674, SFB873/Deutsche Forschungsgemeinschaft (German Research Foundation)
- "Integrate-TN" Consortium/Deutsche Krebshilfe (German Cancer Aid)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
