

Functional profiling of murine glioma models highlights targetable immune evasion phenotypes
- PMID: 39592459
- PMCID: PMC11599368
- DOI: 10.1007/s00401-024-02831-w
Functional profiling of murine glioma models highlights targetable immune evasion phenotypes
Abstract
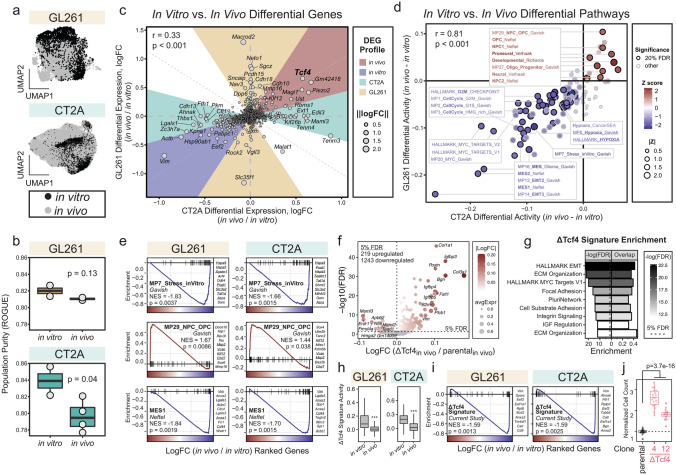
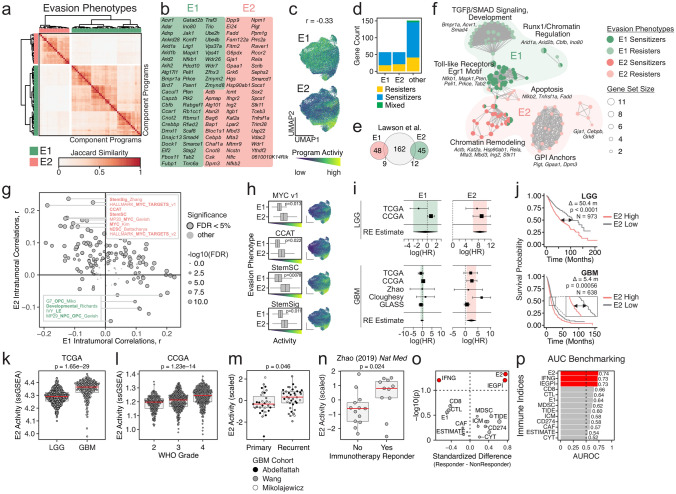
Cancer-intrinsic immune evasion mechanisms and pleiotropy are a barrier to cancer immunotherapy. This is apparent in certain highly fatal cancers, including high-grade gliomas and glioblastomas (GBM). In this study, we evaluated two murine syngeneic glioma models (GL261 and CT2A) as preclinical models for human GBM using functional genetic screens, single-cell transcriptomics and machine learning approaches. Through CRISPR genome-wide co-culture killing screens with various immune cells (cytotoxic T cells, natural killer cells, and macrophages), we identified three key cancer-intrinsic evasion mechanisms: NFκB signaling, autophagy/endosome machinery, and chromatin remodeling. Additional fitness screens identified dependencies in murine gliomas that partially recapitulated those seen in human GBM (e.g., UFMylation). Our single-cell analyses showed that different glioma models exhibited distinct immune infiltration patterns and recapitulated key immune gene programs observed in human GBM, including hypoxia, interferon, and TNF signaling. Moreover, in vivo orthotopic tumor engraftment was associated with phenotypic shifts and changes in proliferative capacity, with murine tumors recapitulating the intratumoral heterogeneity observed in human GBM, exhibiting propensities for developmental- and mesenchymal-like phenotypes. Notably, we observed common transcription factors and cofactors shared with human GBM, including developmental (Nfia and Tcf4), mesenchymal (Prrx1 and Wwtr1), as well as cycling-associated genes (Bub3, Cenpa, Bard1, Brca1, and Mis18bp1). Perturbation of these genes led to reciprocal phenotypic shifts suggesting intrinsic feedback mechanisms that balance in vivo cellular states. Finally, we used a machine-learning approach to identify two distinct immune evasion gene programs, one of which represents a clinically-relevant phenotype and delineates a subpopulation of stem-like glioma cells that predict response to immune checkpoint inhibition in human patients. This comprehensive characterization helps bridge the gap between murine glioma models and human GBM, providing valuable insights for future therapeutic development.
Keywords: CT2A; GL261; Genome-wide CRISPR screen; Glioblastoma; Glioma; Human; Murine; scRNA-seq.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Conflict of interest: The research was conducted in the absence of any commercial/financial relationships that could be construed as a conflict of interest.
Figures






References
-
- Ausman JI, Shapiro WR, Rall DP (1970) Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res 30:2394–2400 - PubMed
-
- Azzarelli B, Miravalle L, Vidal R (2004) Immunolocalization of the oligodendrocyte transcription factor 1 (Olig1) in brain tumors. J Neuropathol Exp Neurol 63:170–179. 10.1093/jnen/63.2.170 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
