

A proteome-wide structural systems approach reveals insights into protein families of all human herpesviruses
- PMID: 39592652
- PMCID: PMC11599850
- DOI: 10.1038/s41467-024-54668-2
A proteome-wide structural systems approach reveals insights into protein families of all human herpesviruses
Abstract
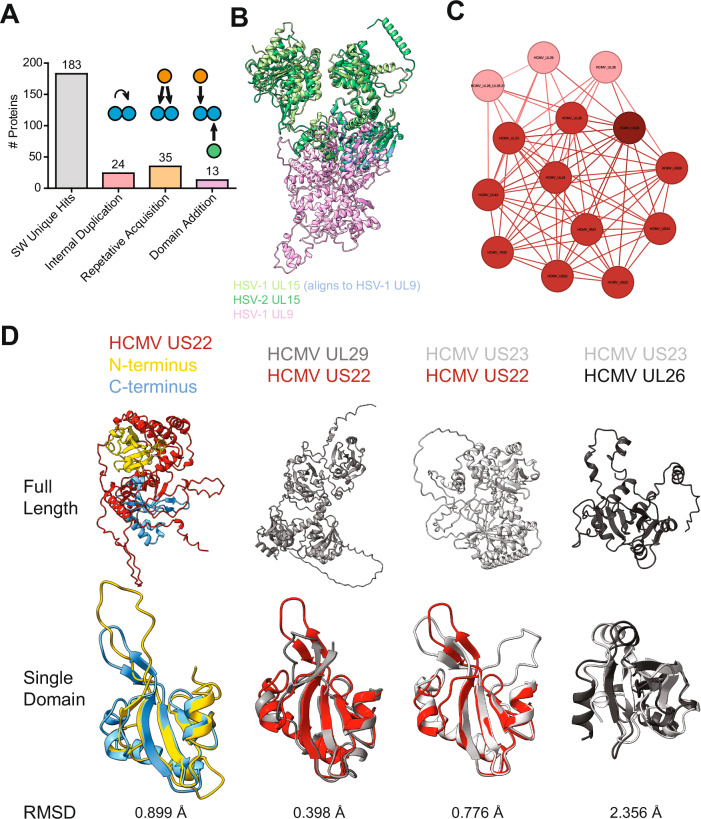
Structure predictions have become invaluable tools, but viral proteins are absent from the EMBL/DeepMind AlphaFold database. Here, we provide proteome-wide structure predictions for all nine human herpesviruses and analyze them in depth with explicit scoring thresholds. By clustering these predictions into structural similarity groups, we identified new families, such as the HCMV UL112-113 cluster, which is conserved in alpha- and betaherpesviruses. A domain-level search found protein families consisting of subgroups with varying numbers of duplicated folds. Using large-scale structural similarity searches, we identified viral proteins with cellular folds, such as the HSV-1 US2 cluster possessing dihydrofolate reductase folds and the EBV BMRF2 cluster that might have emerged from cellular equilibrative nucleoside transporters. Our HerpesFolds database is available at https://www.herpesfolds.org/herpesfolds and displays all models and clusters through an interactive web interface. Here, we show that system-wide structure predictions can reveal homology between viral species and identify potential protein functions.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
- FOR5200 DEEP-DV (443644894) project KA 3492/12-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- Leibniz ScienceCampus InterACt/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- WT_/Wellcome Trust/United Kingdom
- Collaborative Award (209250/Z/17/Z)/Wellcome Trust (Wellcome)
- W75/2022 InterACt/Leibniz-Gemeinschaft (Leibniz Association)
LinkOut - more resources
Full Text Sources
