

Understanding the biological processes of kidney carcinogenesis: an integrative multi-omics approach
- PMID: 39592856
- PMCID: PMC11612429
- DOI: 10.1038/s44320-024-00072-3
Understanding the biological processes of kidney carcinogenesis: an integrative multi-omics approach
Abstract
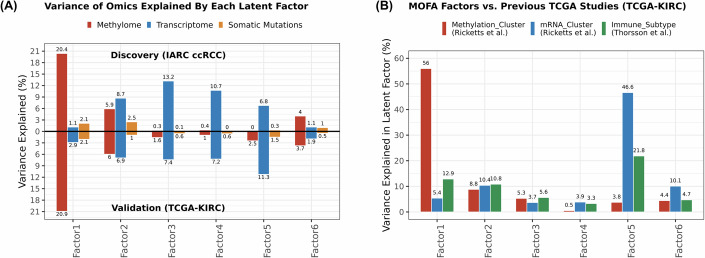
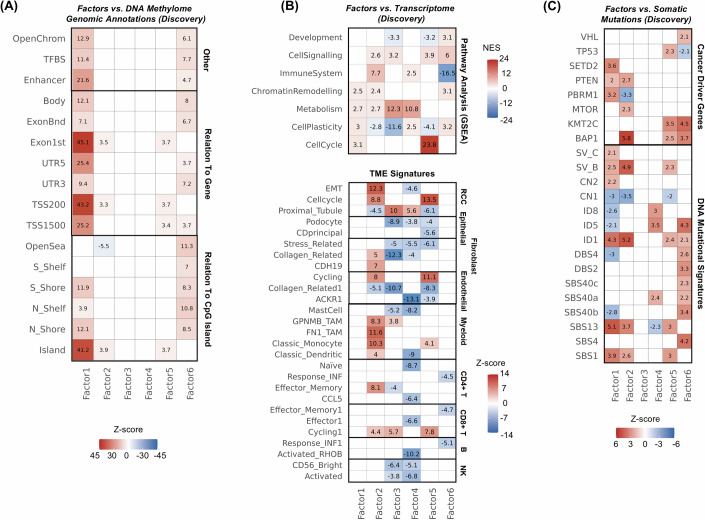
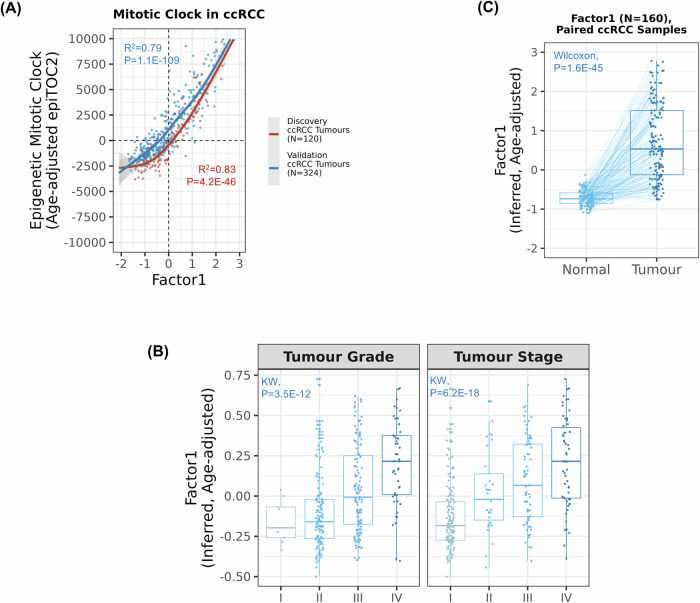
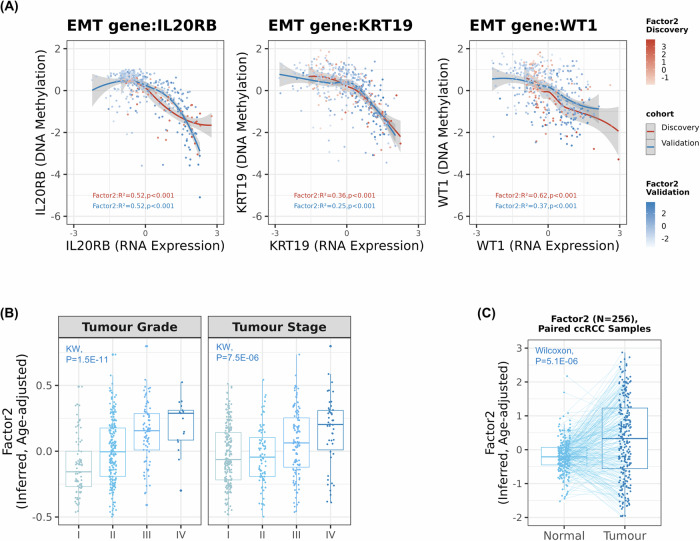
Biological mechanisms related to cancer development can leave distinct molecular fingerprints in tumours. By leveraging multi-omics and epidemiological information, we can unveil relationships between carcinogenesis processes that would otherwise remain hidden. Our integrative analysis of DNA methylome, transcriptome, and somatic mutation profiles of kidney tumours linked ageing, epithelial-mesenchymal transition (EMT), and xenobiotic metabolism to kidney carcinogenesis. Ageing process was represented by associations with cellular mitotic clocks such as epiTOC2, SBS1, telomere length, and PBRM1 and SETD2 mutations, which ticked faster as tumours progressed. We identified a relationship between BAP1 driver mutations and the epigenetic upregulation of EMT genes (IL20RB and WT1), correlating with increased tumour immune infiltration, advanced stage, and poorer patient survival. We also observed an interaction between epigenetic silencing of the xenobiotic metabolism gene GSTP1 and tobacco use, suggesting a link to genotoxic effects and impaired xenobiotic metabolism. Our pan-cancer analysis showed these relationships in other tumour types. Our study enhances the understanding of kidney carcinogenesis and its relation to risk factors and progression, with implications for other tumour types.
Keywords: Cancer Biology; Genomic Epidemiology; Integrative Multi-omics Analysis; Kidney Cancer; Tumour Microenvironment.
© 2024. World Health Organization.
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policies, or views of the International Agency for Research on Cancer/World Health Organization. This study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki. Informed consent was obtained for all participants included in the discovery and validation sets. Ethical approvals were obtained from Local and Federal Research Ethics Committees, and from the IARC Ethics Committee (IEC Project 17-10A4). For the TCGA datasets, also used as validation set, the enrolling, collection, clinical and genomic data processing and distributions are subject to 45-CFR-46 (the “Common Rule”) governing protection of human research subjects. Under the revised TCGA consent policy, re-consent of still-living participants is no longer a programme-imposed requirement. The Project Team described the best practices for informed consent for participating in TCGA in this memo ( http://cancergenome.nih.gov/abouttcga/policies/informedconsent ).
Figures






References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
