

Novel PP2A-Activating Compounds in Neuroblastoma
- PMID: 39594793
- PMCID: PMC11592631
- DOI: 10.3390/cancers16223836
Novel PP2A-Activating Compounds in Neuroblastoma
Abstract
Background: Neuroblastoma (NB) remains one of the deadliest pediatric solid tumors. Recent advancements aimed at improving outcomes have been insufficient, and patients with high-risk NB continue to have a poor prognosis. Protein phosphatase 2A (PP2A) is a tumor suppressor protein downregulated in many cancers, including NB. PP2A activation has been shown to affect the malignant phenotype in other solid tumors. The present studies aim to investigate the effects of two novel PP2A activators as a NB therapeutic.
Methods: Four established NB cell lines and a patient-derived xenoline were utilized to study the effect on cell viability, proliferation, motility, and in vivo tumor growth using two novel tricyclic sulfonamide PP2A activators, ATUX-3364 and ATUX-8385.
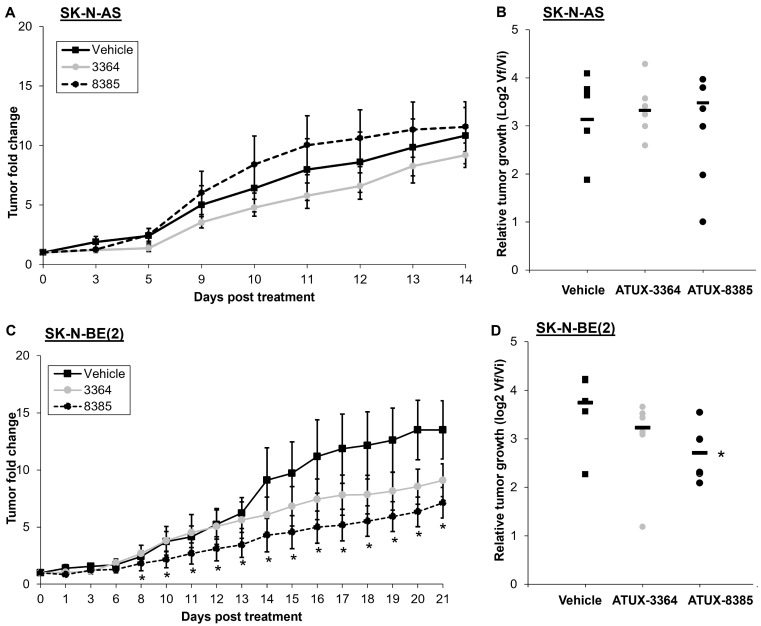
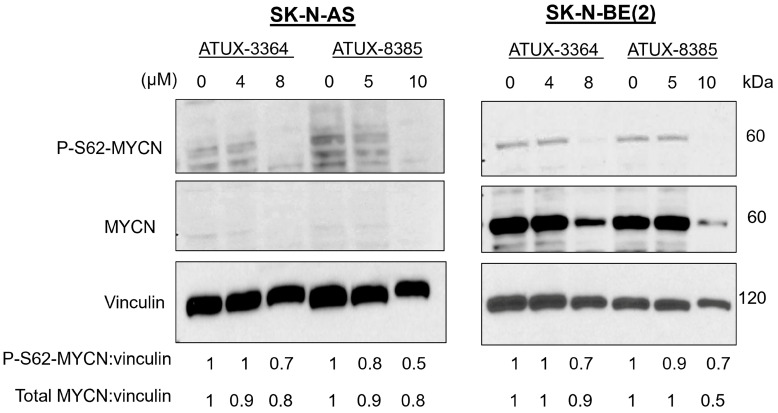
Results: ATUX-3364 and ATUX-8385 increased PP2A activity. These PP2A activators led to decreased viability, proliferation, and motility of NB cells. Treatment of animals bearing NB tumors with ATUX-3364 or ATUX-8385 resulted in decreased tumor growth in MYCN-amplified SK-N-BE(2) tumors. At the molecular level, PP2A-based reactivation led to dephosphorylation of MYCN-S62 and decreased MYCN protein expression.
Conclusions: PP2A activators decreased NB cell viability, proliferation, and motility. In vivo experiments show that PP2A activators have more significant effects on tumorigenesis in MYCN-amplified tumors. Finally, phosphorylation of MYCN protein was decreased following treatment with novel sulfonamide PP2A activators. These data and mechanistic insights may be useful for developing new PP2A-based therapies that target MYCN for the treatment of NB.
Keywords: ATUX-3364; ATUX-8385; neuroblastoma; protein phosphatase 2A.
Conflict of interest statement
M.O. is the inventor of ATUX-3364 and ATUX-8385 and the founder of Atux Iskay, LLC. The other authors declare no competing financial interests in relation to the work described.
Figures
References
-
- Cohn S.L., Pearson A.D.J., London W.B., Monclair T., Ambros P.F., Brodeur G.M., Faldum A., Hero B., Iehara T., Machin D., et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
