

Neutrophils and Lymphocytes: Yin and Yang of Lung Fibrosis and Patient Outcome in Diffuse Interstitial Lung Diseases
- PMID: 39595006
- PMCID: PMC11592343
- DOI: 10.3390/biomedicines12112439
Neutrophils and Lymphocytes: Yin and Yang of Lung Fibrosis and Patient Outcome in Diffuse Interstitial Lung Diseases
Abstract
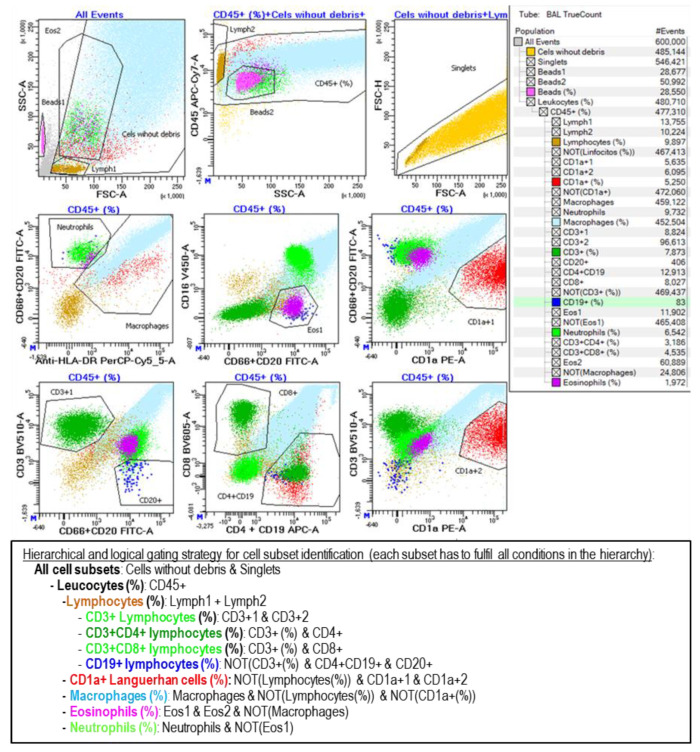
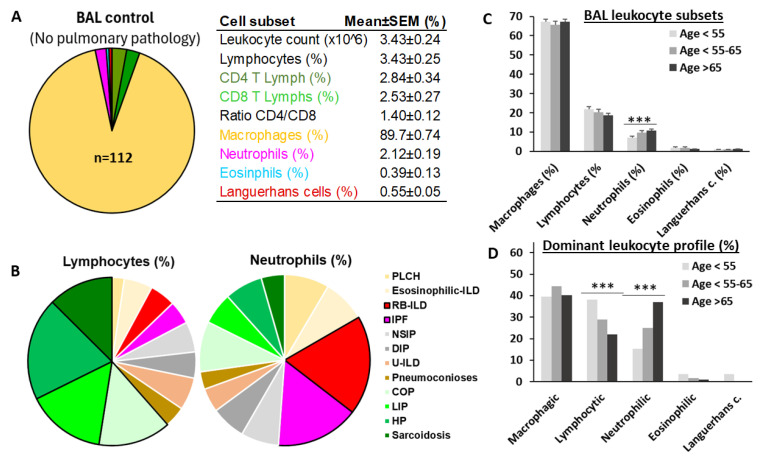
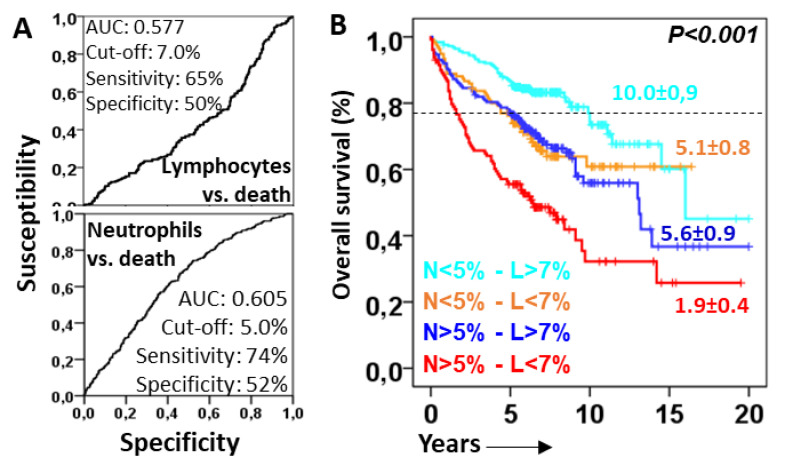
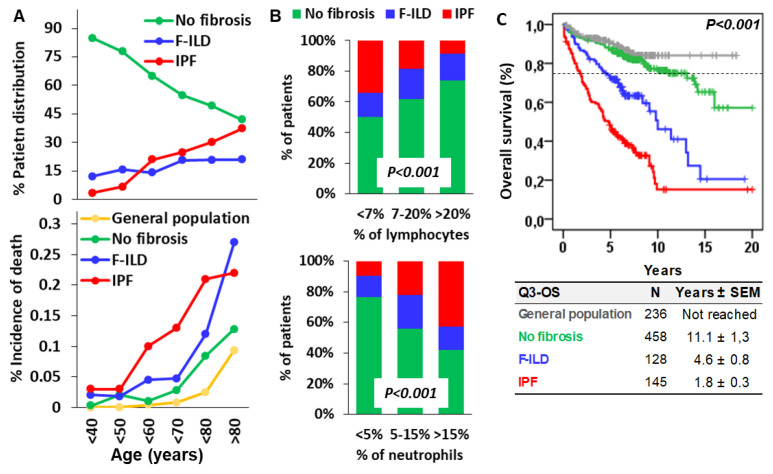
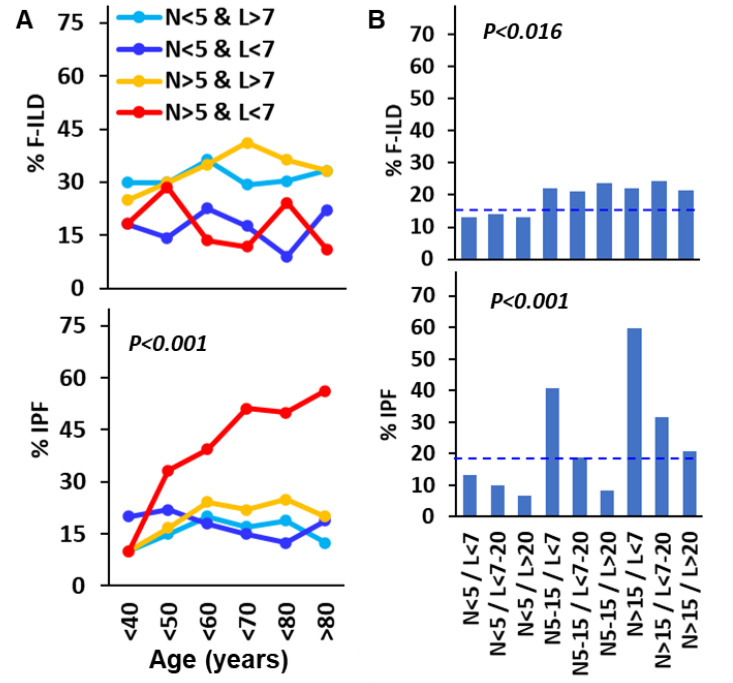
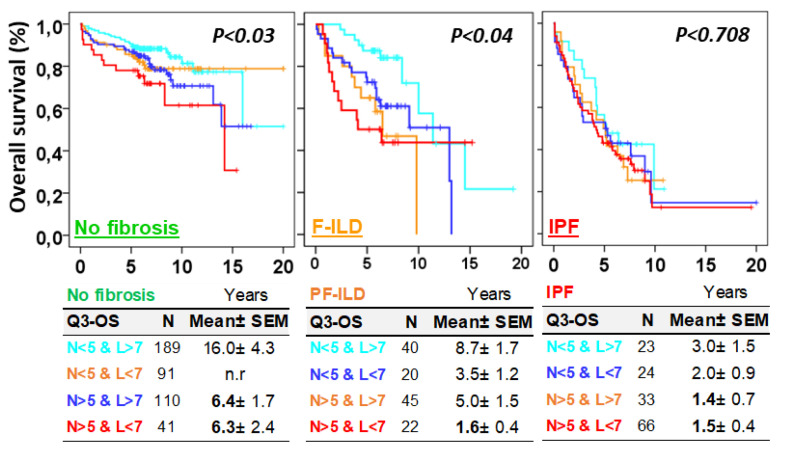
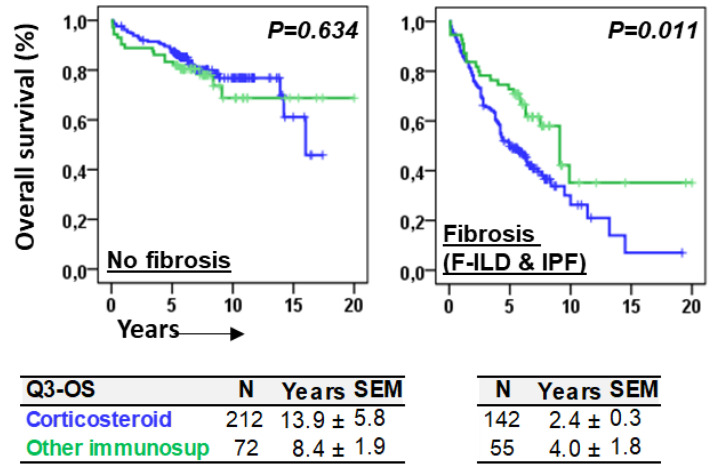
Objective: Antifibrotics can improve the outcome of patients with idiopathic pulmonary fibrosis (IPF) and other fibrosing interstitial lung diseases (F-ILDs), but predictive biomarkers at diagnosis are needed to guide the use of immunomodulating and antifibrotic therapies. Methods: Flow cytometry quantification of lymphocytes and neutrophils in bronchoalveolar lavage (BAL) of 145 IPFs, 561 non-IPF-ILDs (125 F-ILDs), and 112 BAL controls were retrospectively correlated with the incidence of fibrosis and third-quartile overall survival (Q3-OS). Results: The incidence of IPF was directly proportional (9.6%, 22.2%, and 42.6%, p < 0.001) to BAL neutrophil counts (<5%, 5-15%, and >15%), but inversely proportional (34.1%, 18.6%, and 8.8%, p < 0.001) to BAL lymphocyte counts (<7%, 7-20%, and >20%). Elevated neutrophils (>5%) with low lymphocytes (<7%) were associated with an increasingly higher incidence of IPF (10.0-56.3%, p < 0.001) in patients aged 40 to 80, compared to the rest of patients (13.0-17.1%). Lymphocytes >20% compared to lymphocytes <7% strongly protected patients with neutrophils >15% (59.7% vs. 20.7%, p < 0.001) from IPF. In contrast, the incidence of F-ILD was not clearly related to BAL lymphocyte/neutrophil counts. Although, IPF and F-ILD showed a shorter Q3-OS (1.8 ± 0.3 and 4.6 ± 0.8 years; p < 0.001) than non-fibrotic-ILDs (11.1 ± 1.3 years), lymphocyte and neutrophil counts were associated with a longer and shorter Q3-OS of non-fibrotic-ILDs (p < 0.03) and F-ILDs (p < 0.04), respectively, but not with a Q3-OS of IPF patients (p < 0.708). Corticosteroids in patients with fibrosis showed a shorter Q3-OS than other immunomodulators (2.4 ± 0.3 vs. 4.0 ± 1.8 years, p = 0.011). Conclusions: Accurate counting of BAL lymphocytes and neutrophils by flow cytometry in ILD patients at diagnosis could help guide immunomodulatory and antifibrotic therapies.
Keywords: BAL lymphocyte and neutrophils; flow cytometry; idiopathic pulmonary fibrosis; interstitial lung disease; lung fibrosis; patient outcome.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Cottin V., Hirani N.A., Hotchkin D.L., Nambiar A.M., Ogura T., Otaola M., Skowasch D., Park J.S., Poonyagariyagorn H.K., Wuyts W., et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018;27:180076. doi: 10.1183/16000617.0076-2018. - DOI - PMC - PubMed
-
- Wells A.U., Flaherty K.R., Brown K.K., Inoue Y., Devaraj A., Richeldi L., Moua T., Crestani B., Wuyts W.A., Stowasser S., et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases—Subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020;8:453–460. doi: 10.1016/S2213-2600(20)30036-9. - DOI - PubMed
LinkOut - more resources
Full Text Sources
