

Copy Number Variants in Cardiac Channelopathies: Still a Missed Part in Routine Arrhythmic Diagnostics
- PMID: 39595626
- PMCID: PMC11592175
- DOI: 10.3390/biom14111450
Copy Number Variants in Cardiac Channelopathies: Still a Missed Part in Routine Arrhythmic Diagnostics
Abstract
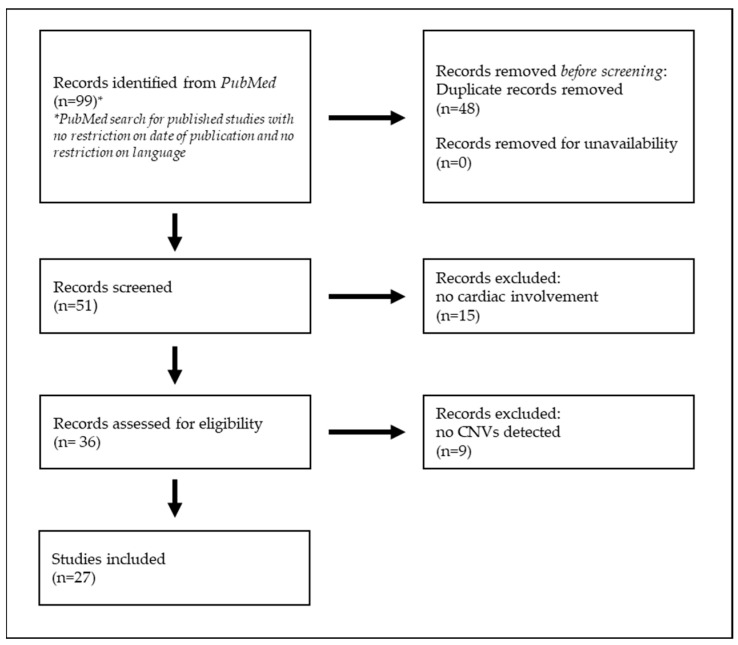
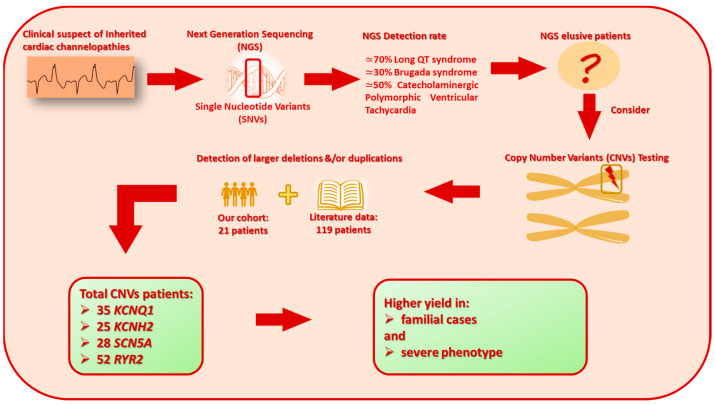
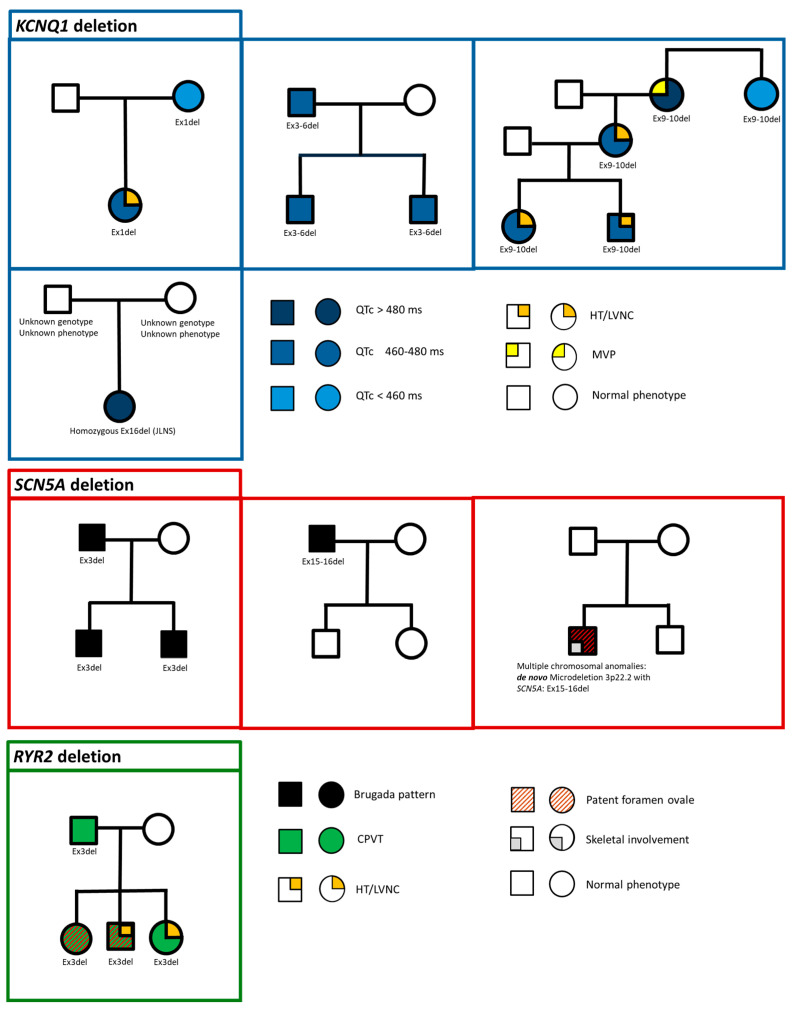
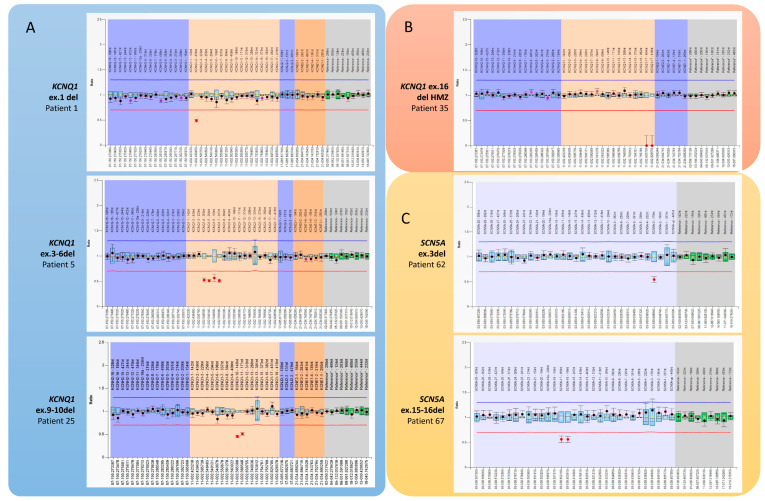
Inherited cardiac channelopathies are major causes of sudden cardiac death (SCD) in young people. Genetic testing is focused on the identification of single-nucleotide variants (SNVs) by Next-Generation Sequencing (NGS). However, genetically elusive cases can carry copy number variants (CNVs), which need specific detection tools. We underlie the utility of identifying CNVs by investigating the literature data and internally analyzing cohorts with CNVs in KCNQ1, KCNH2, SCN5A, and RYR2. CNVs were reported in 119 patients from the literature and 21 from our cohort. Young patients with CNVs in KCNQ1 show a Long QT (LQT) phenotype > 480 ms and a higher frequency of syncope. None of them had SCD. All patients with CNV in KCNH2 had a positive phenotype for QT > 480 ms. CNVs in SCN5A were represented by the Brugada pattern, with major cardiac events mainly in males. Conversely, adult females show more supraventricular arrhythmias. RYR2-exon3 deletion showed a broader phenotype, including left ventricular non-compaction (LVNC) and catecholaminergic polymorphic ventricular tachycardia (CPVT). Pediatric patients showed atrial arrhythmias and paroxysmal atrial fibrillation. Relatively higher syncope and SCA were observed in young females. The detection of CNVs can be of greater yield in two groups: familial channelopathies and patients with suspected Jervell and Lange-Nielsen syndrome or CPVT. The limited number of reported individuals makes it mandatory for multicentric studies to give future conclusive results.
Keywords: Brugada; catecholaminergic polymorphic ventricular tachycardia; channelopathies; copy number variants; long QT.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Priori S.G., Wilde A.A., Horie M., Cho Y., Behr E.R., Berul C., Blom N., Brugada J., Chiang C.-E., Huikuri H., et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document Endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.05.014. - DOI - PubMed
-
- Hayesmoore J.B., Bhuiyan Z.A., Coviello D.A., du Sart D., Edwards M., Iascone M., Morris-Rosendahl D.J., Sheils K., van Slegtenhorst M., Thomson K.L. EMQN: Recommendations for Genetic Testing in Inherited Cardiomyopathies and Arrhythmias. Eur. J. Hum. Genet. 2023;31:1003–1009. doi: 10.1038/s41431-023-01421-w. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
