

Alzheimer's Disease, Obesity, and Type 2 Diabetes: Focus on Common Neuroglial Dysfunctions (Critical Review and New Data on Human Brain and Models)
- PMID: 39595866
- PMCID: PMC11591712
- DOI: 10.3390/brainsci14111101
Alzheimer's Disease, Obesity, and Type 2 Diabetes: Focus on Common Neuroglial Dysfunctions (Critical Review and New Data on Human Brain and Models)
Abstract
Background/objectives: Obesity, type 2 diabetes (T2D), and Alzheimer's disease (AD) are pathologies that affect millions of people worldwide. They have no effective therapy and are difficult to prevent and control when they develop. It has been known for many years that these diseases have many pathogenic aspects in common. We highlight in this review that neuroglial cells (astroglia, oligodendroglia, and microglia) play a vital role in the origin, clinical-pathological development, and course of brain neurodegeneration. Moreover, we include the new results of a T2D-AD mouse model (APP+PS1 mice on a high-calorie diet) that we are investigating.
Methods: Critical bibliographic revision and biochemical neuropathological study of neuroglia in a T2D-AD model.
Results: T2D and AD are not only "connected" by producing complex pathologies in the same individual (obesity, T2D, and AD), but they also have many common pathogenic mechanisms. These include insulin resistance, hyperinsulinemia, hyperglycemia, oxidative stress, mitochondrial dysfunction, and inflammation (both peripheral and central-or neuroinflammation). Cognitive impairment and AD are the maximum exponents of brain neurodegeneration in these pathological processes. both due to the dysfunctions induced by metabolic changes in peripheral tissues and inadequate neurotoxic responses to changes in the brain. In this review, we first analyze the common pathogenic mechanisms of obesity, T2D, and AD (and/or cerebral vascular dementia) that induce transcendental changes and responses in neuroglia. The relationships between T2D and AD discussed mainly focus on neuroglial responses. Next, we present neuroglial changes within their neuropathological context in diverse scenarios: (a) aging involution and neurodegenerative disorders, (b) human obesity and diabetes and obesity/diabetes models, (c) human AD and in AD models, and (d) human AD-T2D and AD-T2D models. An important part of the data presented comes from our own studies on humans and experimental models over the past few years. In the T2D-AD section, we included the results of a T2D-AD mouse model (APP+PS1 mice on a high-calorie diet) that we investigated, which showed that neuroglial dysfunctions (astrocytosis and microgliosis) manifest before the appearance of amyloid neuropathology, and that the amyloid pathology is greater than that presented by mice fed a normal, non-high-caloric diet A broad review is finally included on pharmacological, cellular, genic, and non-pharmacological (especially diet and lifestyle) neuroglial-related treatments, as well as clinical trials in a comparative way between T2D and AD. These neuroglial treatments need to be included in the multimodal/integral treatments of T2D and AD to achieve greater therapeutic efficacy in many millions of patients.
Conclusions: Neuroglial alterations (especially in astroglia and microglia, cornerstones of neuroinflammation) are markedly defining brain neurodegeneration in T2D and A, although there are some not significant differences between each of the studied pathologies. Neuroglial therapies are a very important and p. promising tool that are being developed to prevent and/or treat brain dysfunction in T2D-AD. The need for further research in two very different directions is evident: (a) characterization of the phenotypic changes of astrocytes and microglial cells in each region of the brain and in each phase of development of each isolated and associated pathology (single-cell studies are mandatory) to better understand the pathologies and define new therapeutic targets; (b) studying new therapeutic avenues to normalize the function of neuroglial cells (preventing neurotoxic responses and/or reversing them) in these pathologies, as well as the phenotypic characteristics in each moment of the course and place of the neurodegenerative process.
Keywords: Alzheimer’s disease (AD); astroglia; microglia; neuroglia; neuroglial dysfunctions; neuroglial therapy; neuroinflammation; neuropathology; obesity; oligodendroglia; pathogenic mechanisms; type 2 diabetes (T2D).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

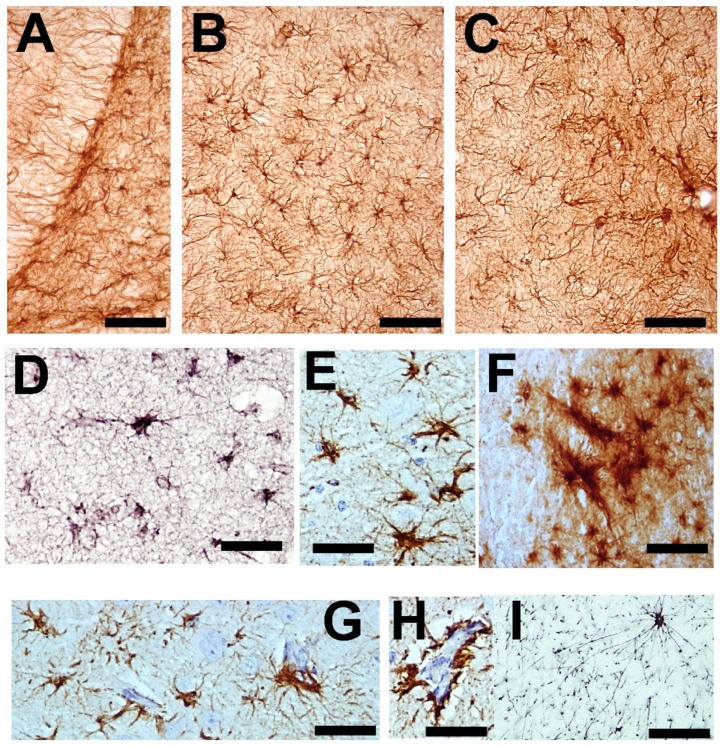
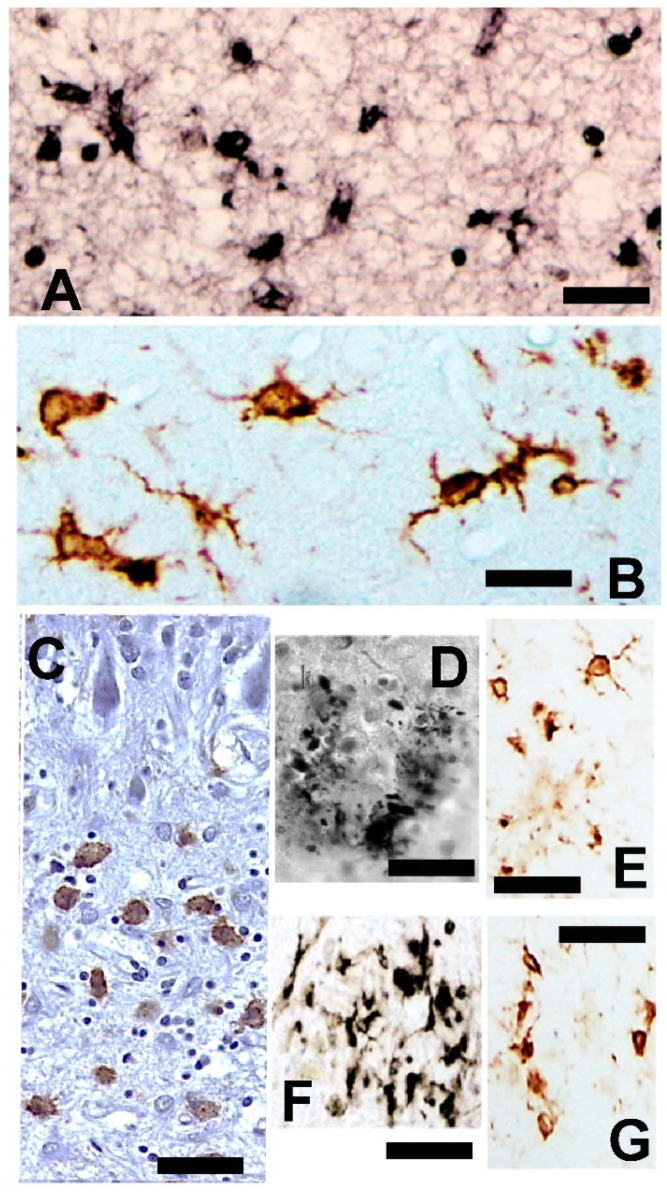
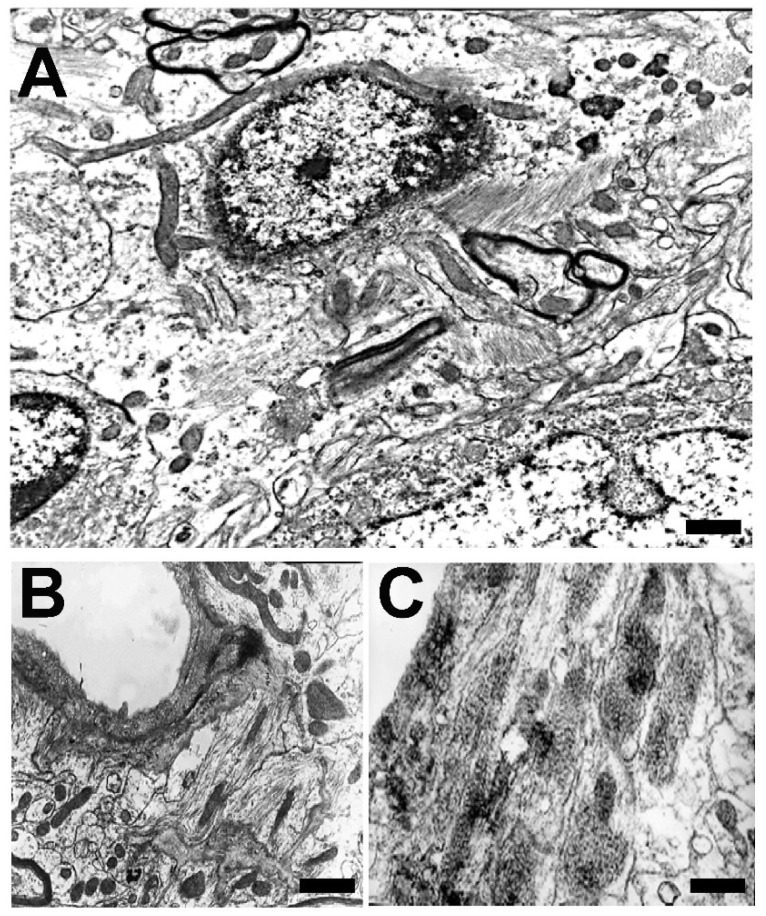
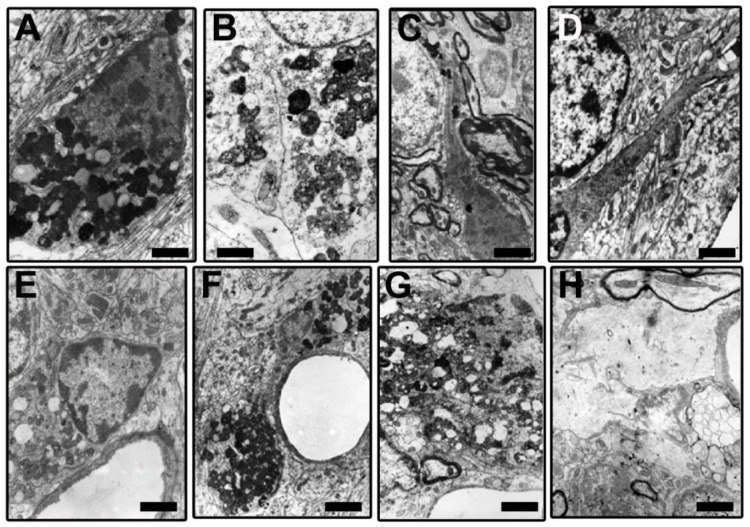
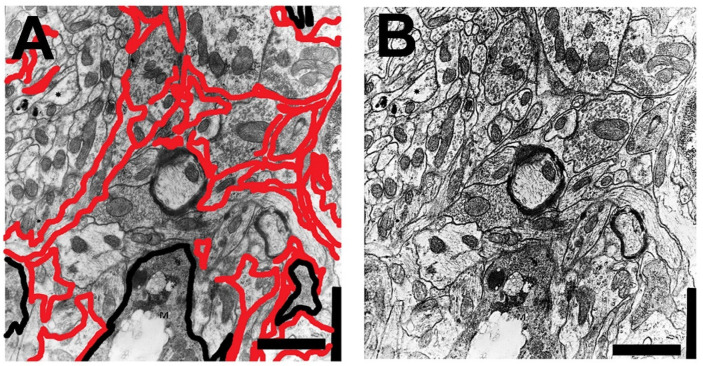
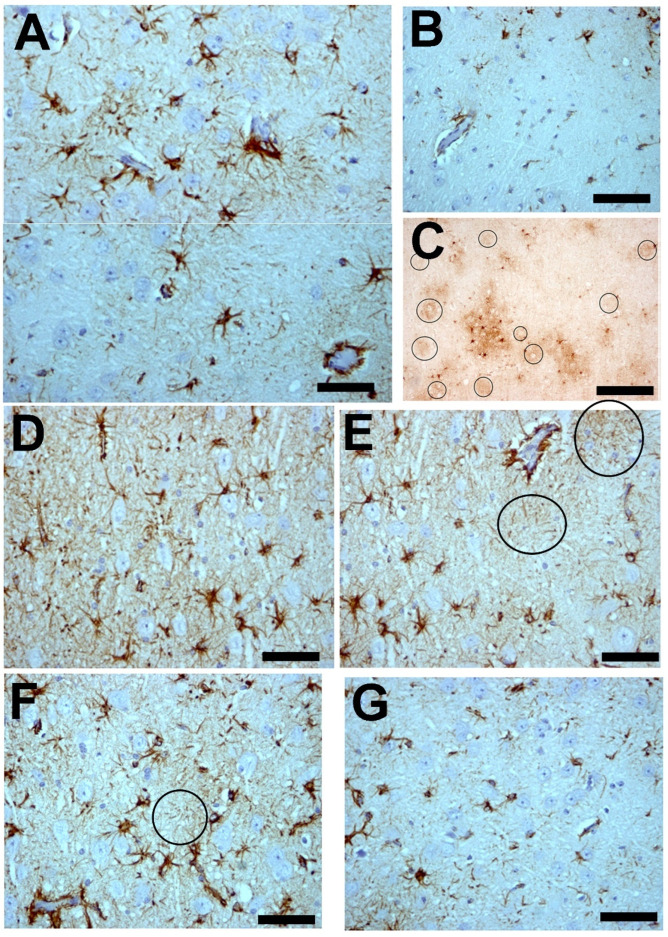

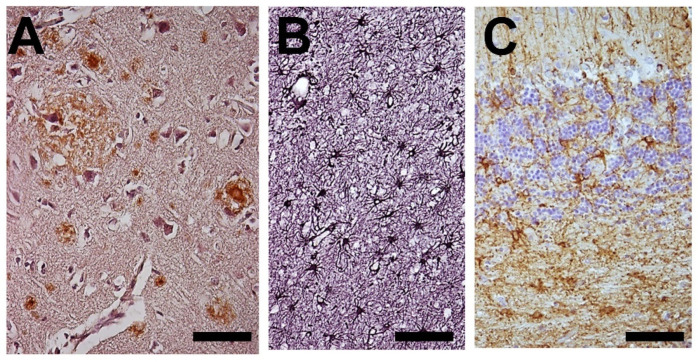
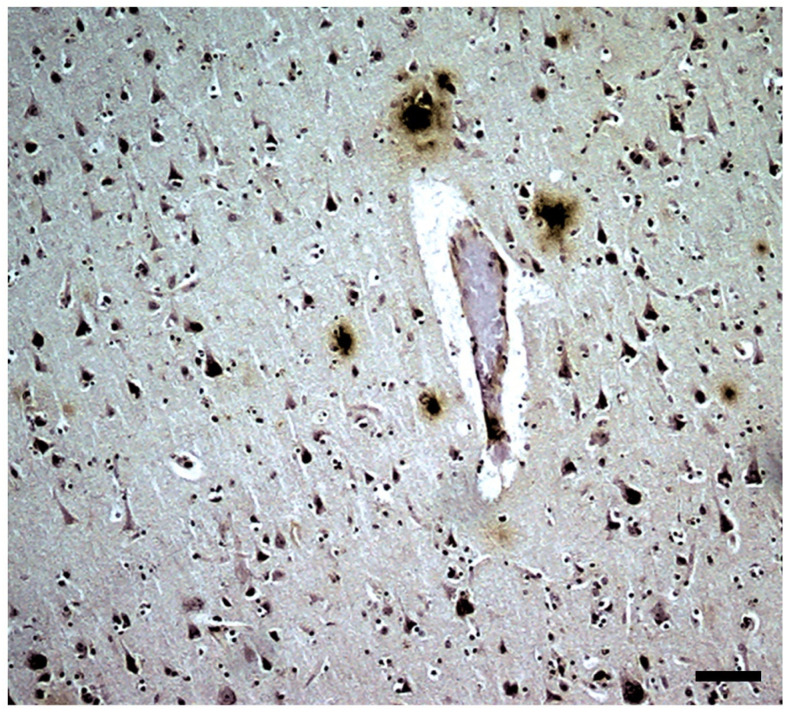


 ) or hypercaloric (H; blue–white bars
) or hypercaloric (H; blue–white bars  ) diet versus the mean values given by wild type (non-transgenic) controls. Cholesterol concentrations in the liver (mg/g liver tissue) (Chol liver) are also shown to show damage to peripheral tissues (increasing “metabolic disease” in peripheral organs), which may secondarily increase neuroinflammation. In the groups treated with a hyper-caloric diet, there are statistically significant differences versus the normal control (asterisk) and statistically significant differences versus genetically modified mice fed a normal diet (black square).
) diet versus the mean values given by wild type (non-transgenic) controls. Cholesterol concentrations in the liver (mg/g liver tissue) (Chol liver) are also shown to show damage to peripheral tissues (increasing “metabolic disease” in peripheral organs), which may secondarily increase neuroinflammation. In the groups treated with a hyper-caloric diet, there are statistically significant differences versus the normal control (asterisk) and statistically significant differences versus genetically modified mice fed a normal diet (black square).

References
-
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 5th ed. American Psychiatric Association; Arlington, VA, USA: 2013. TR.
-
- McKhann G.M., Knopman D.S., Chertkow H. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
