

In Vitro Gene Therapy Using Human iPS-Derived Mesoangioblast-Like Cells (HIDEMs) Combined with Microdystrophin (μDys) Expression as the New Strategy for Duchenne Muscular Dystrophy (DMD) Experimental Treatment
- PMID: 39595938
- PMCID: PMC11593506
- DOI: 10.3390/ijms252211869
In Vitro Gene Therapy Using Human iPS-Derived Mesoangioblast-Like Cells (HIDEMs) Combined with Microdystrophin (μDys) Expression as the New Strategy for Duchenne Muscular Dystrophy (DMD) Experimental Treatment
Abstract
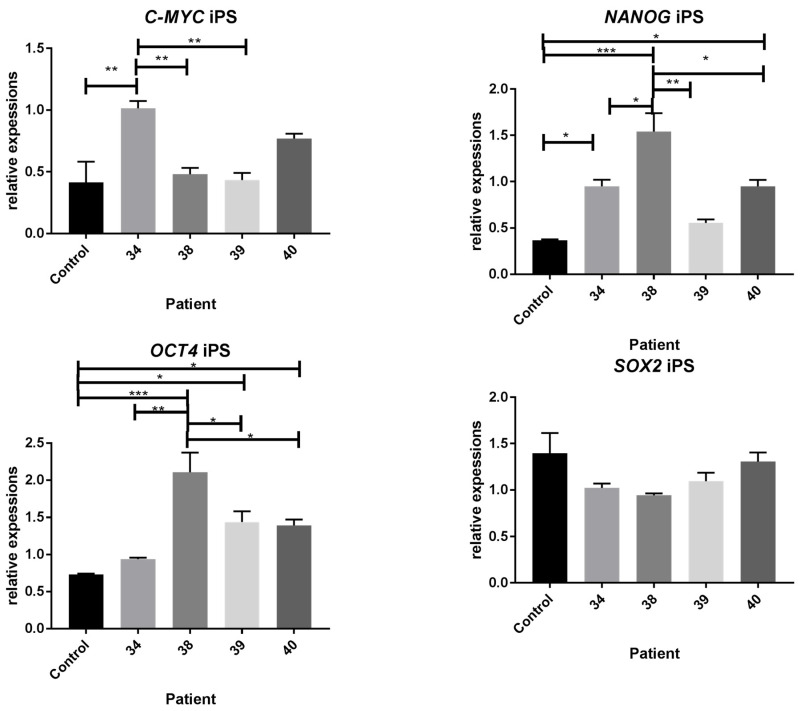
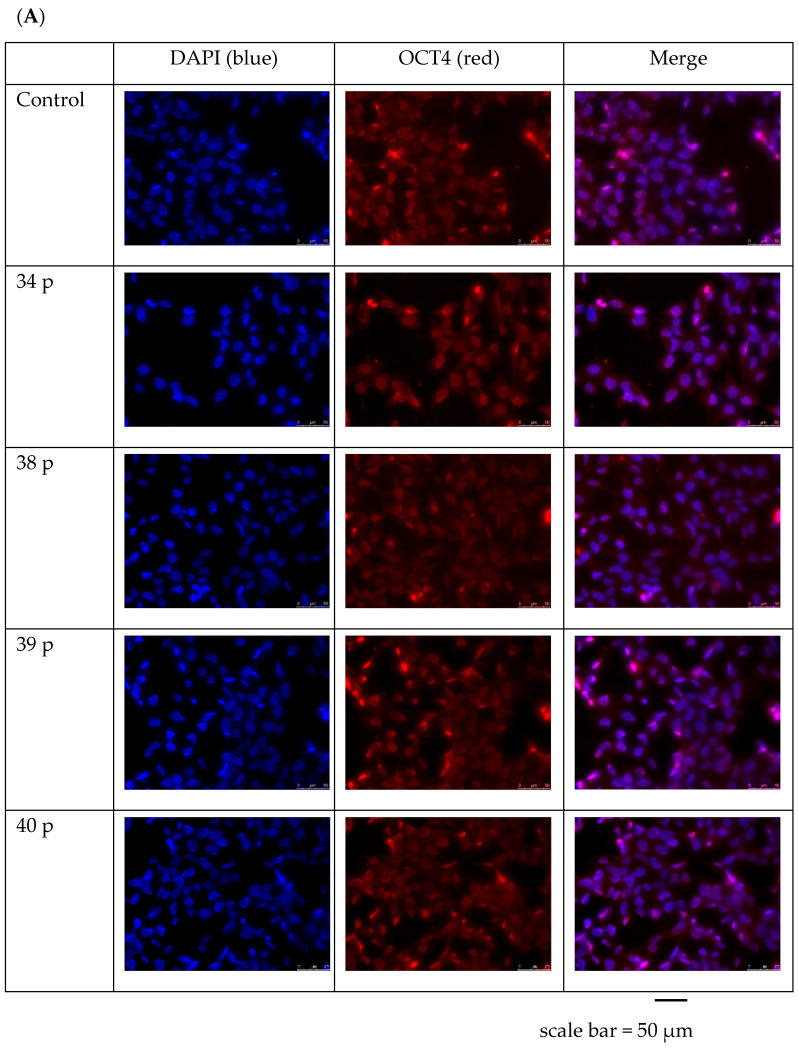
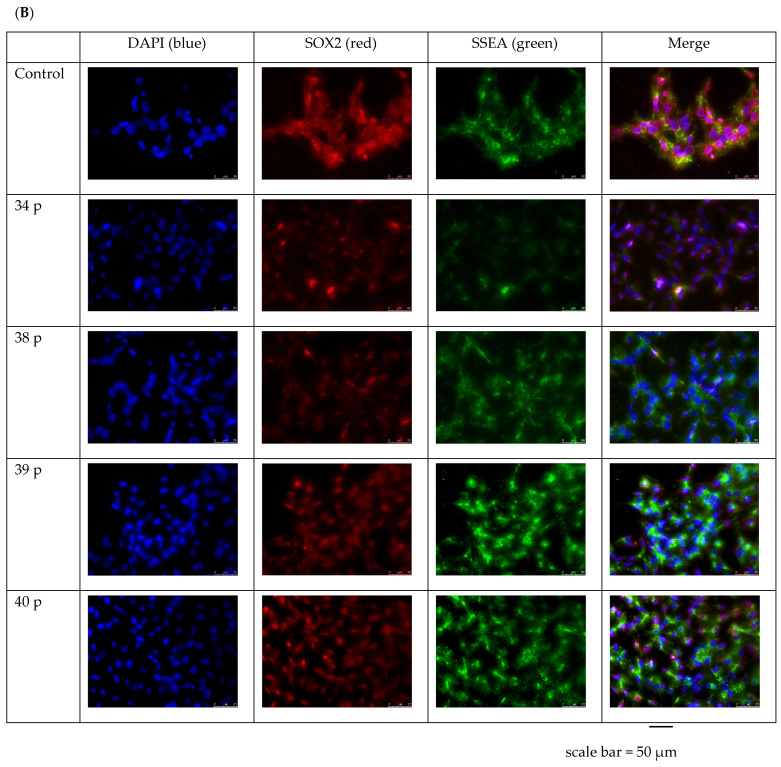
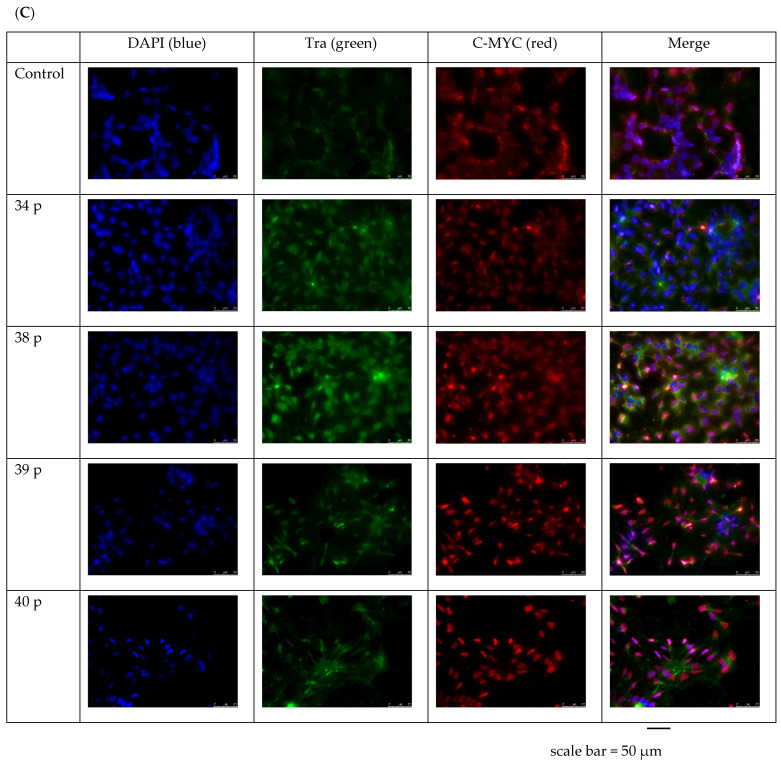
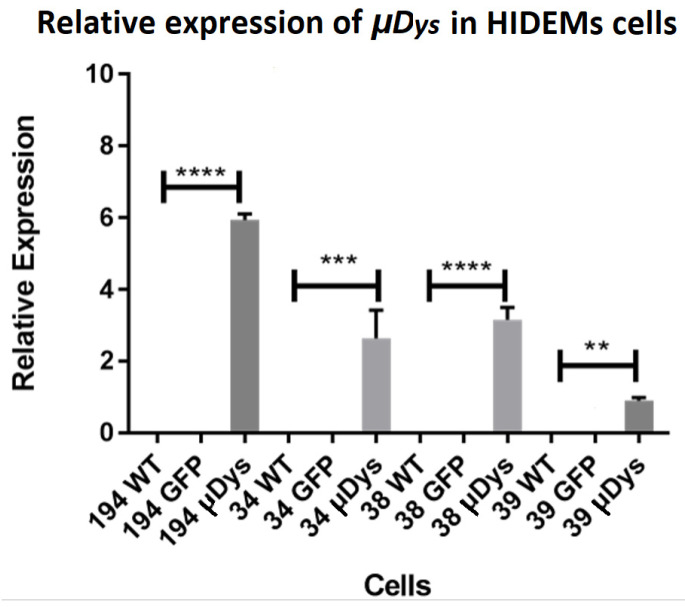
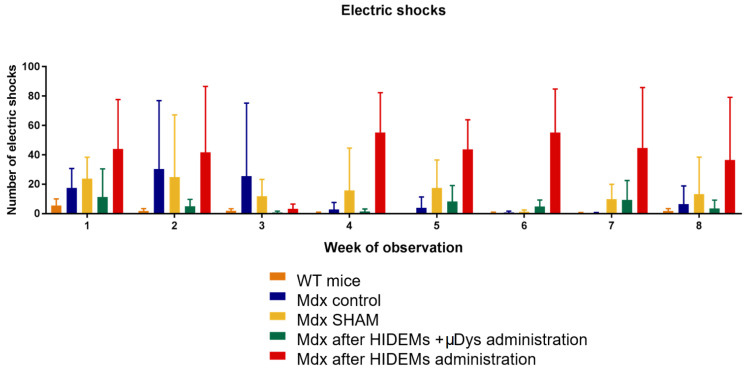
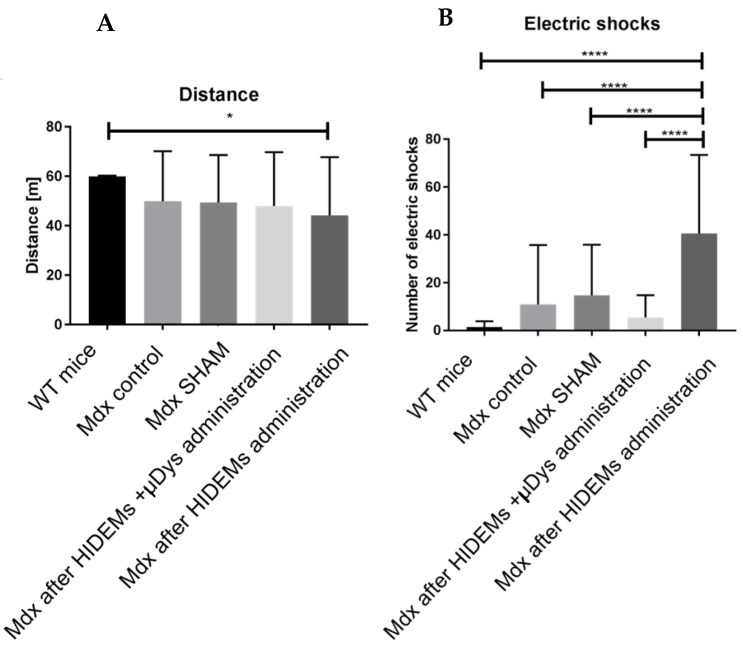
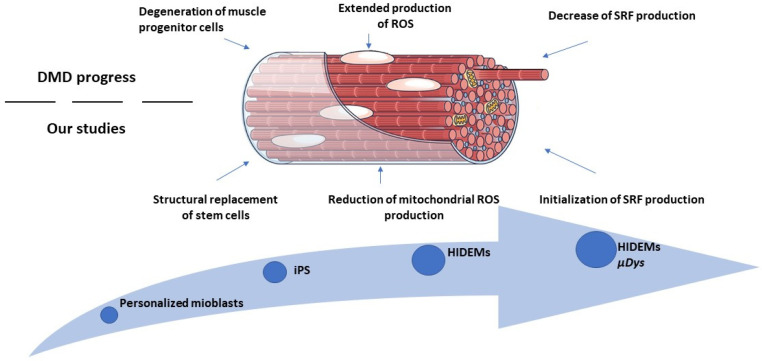
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by disruptions in the dystrophin gene. This study aims to investigate potential a therapeutic approach using genetically modified human iPS-derived mesoangioblast-like cells (HIDEMs) in mdx mouse model. This study utilizes patient-specific myoblasts reprogrammed to human induced pluripotent stem cells (iPSCs) and then differentiated into HIDEMs. Lentiviral vectors carrying microdystrophin sequences have been employed to deliver the genetic construct to express a shortened, functional dystrophin protein in HIDEMs. The study indicated significant changes within redox potential between healthy and pathological HIDEM cells derived from DMD patients studied by catalase and superoxide dismutase activities. Microdystrophin expressing HIDEMs also improved expression of genes involved in STARS (striated muscle activator of Rho signaling) pathway albeit in selective DMD patients (with mild phenotype). Although in vivo observations did not bring progress in the mobility of mdx mice with HIDEMs, microdystrophin interventions this may argue against "treadmill test" as suitable for assessment of mdx mice recovery. Low-level signaling of the Rho pathway and inflammation-related factors in DMD myogenic cells can also contribute to the lack of success in a functional study. Overall, this research contributes to the understanding of DMD pathogenesis and provides insights into potential novel therapeutic strategy, highlighting the importance of personalized gene therapy.
Keywords: Duchenne Muscular Dystrophy; gene therapy; mdx mice; microdystrophin; muscular dystrophy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


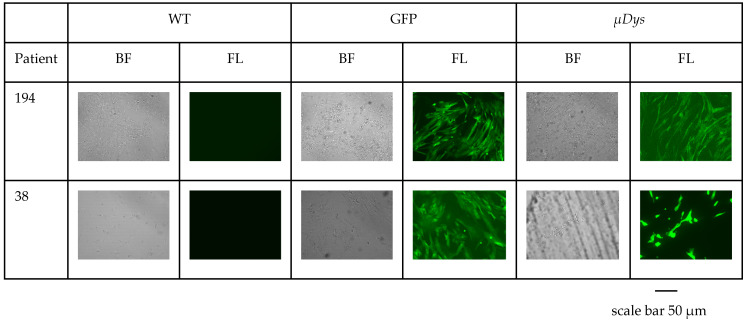
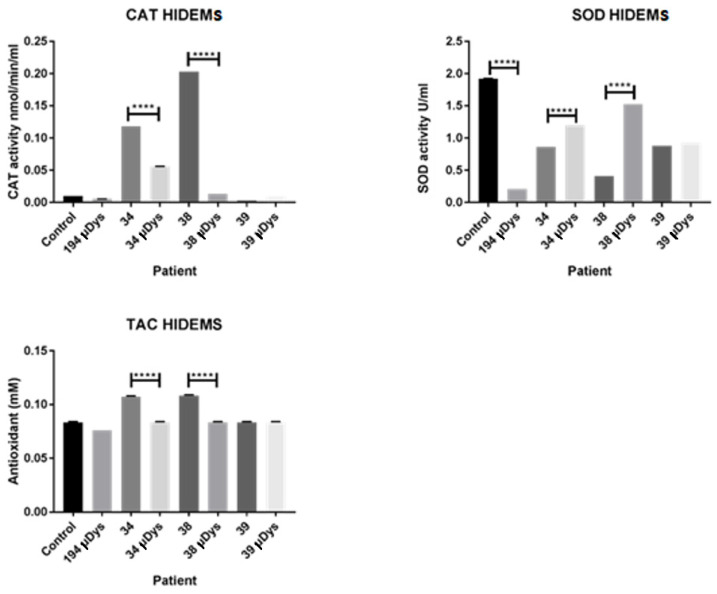
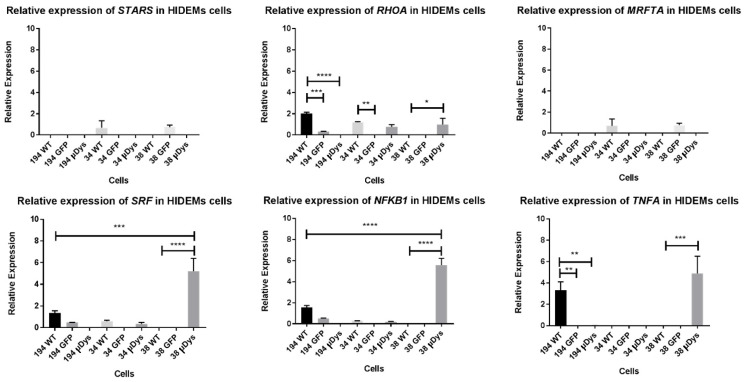
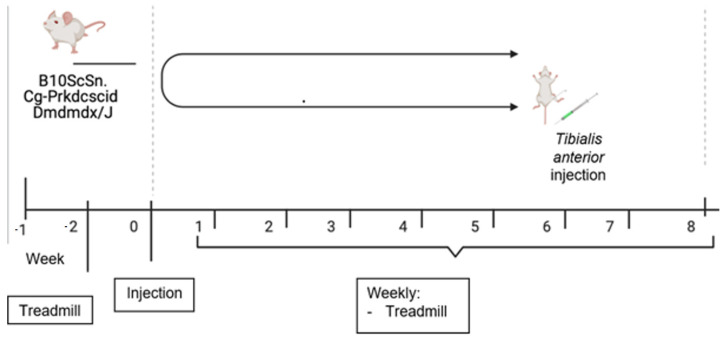
References
-
- Bushby K.M.D., Gardner-Medwin D., Nicholson L.V.B., Johnson M.A., Haggerty I.D., Cleghorn N.J., Harris J.B., Bhattacharyal S.S. The Clinical, Genetic and Dystrophin Characteristics of Becker Muscular Dystrophy—II. Correlation of Phenotype with Genetic and Protein Abnormalities. J. Neurol. 1993;240:105–112. doi: 10.1007/BF00858726. - DOI - PubMed
-
- Van Putten M., Hulsker M., Young C., Nadarajah V.D., Heemskerk H., Van Der Weerd L., ’T Hoen P.A.C., Van Ommen G.J.B., Aartsma-Rus A.M. Low Dystrophin Levels Increase Survival and Improve Muscle Pathology and Function in Dystrophin/Utrophin Double-Knockout Mice. FASEB J. 2013;27:2484–2495. doi: 10.1096/fj.12-224170. - DOI - PMC - PubMed
-
- McDonald C.M., Marbán E., Hendrix S., Hogan N., Ruckdeschel Smith R., Eagle M., Finkel R.S., Tian C., Janas J., Harmelink M.M., et al. Repeated Intravenous Cardiosphere-Derived Cell Therapy in Late-Stage Duchenne Muscular Dystrophy (HOPE-2): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet. 2022;399:1049–1058. doi: 10.1016/S0140-6736(22)00012-5. - DOI - PubMed
-
- Komaki H., Nagata T., Saito T., Masuda S., Takeshita E., Sasaki M., Tachimori H., Nakamura H., Aoki Y., Takeda S. Systemic Administration of the Antisense Oligonucleotide NS-065/NCNP-01 for Skipping of Exon 53 in Patients with Duchenne Muscular Dystrophy. Sci. Transl. Med. 2018;10:eaan0713. doi: 10.1126/scitranslmed.aan0713. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
