

The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies
- PMID: 39596103
- PMCID: PMC11593457
- DOI: 10.3390/ijms252212034
The Role of Human-Induced Pluripotent Stem Cells in Studying Cardiac Channelopathies
Abstract
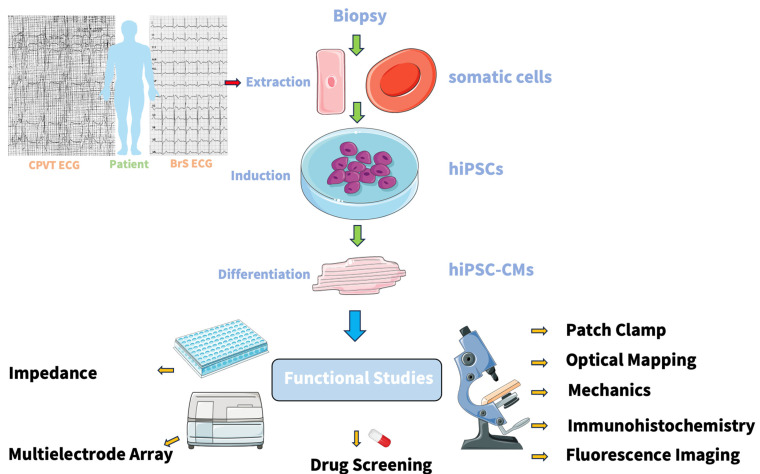
Cardiac channelopathies are inherited diseases that increase the risk of sudden cardiac death. While different genes have been associated with inherited channelopathies, there are still subtypes, e.g., catecholaminergic polymorphic ventricular tachycardia and Brugada syndrome, where the genetic cause remains unknown. Various models, including animal models, heterologous expression systems, and the human-induced pluripotent stem-cell-derived cardiomyocytes (hiPSCs-CMs) model, have been used to study the pathophysiological mechanisms of channelopathies. Recently, researchers have focused on using hiPSCs-CMs to understand the genotype-phenotype correlation and screen drugs. By combining innovative techniques such as Clustered Regularly Interspaced Short Palindromic Repeats/Clustered Regularly Interspaced Short Palindromic Repeats associated protein 9 (CRISPR/Cas9)-mediated genome editing, and three-dimensional (3D) engineered heart tissues, we can gain new insights into the pathophysiological mechanisms of channelopathies. This approach holds promise for improving personalized drug treatment. This review highlights the role of hiPSCs-CMs in understanding the pathomechanism of Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia and how these models can be utilized for drug screening.
Keywords: Brugada syndrome; catecholaminergic polymorphic ventricular tachycardia; sudden cardiac death.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Catecholaminergic Polymorphic Ventricular Tachycardia: Advancing From Molecular Insights to Preclinical Models.J Am Heart Assoc. 2025 Mar 18;14(6):e038308. doi: 10.1161/JAHA.124.038308. Epub 2025 Mar 13. J Am Heart Assoc. 2025. PMID: 40079282 Free PMC article. Review.
-
Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R.Heart Rhythm. 2021 Feb;18(2):250-260. doi: 10.1016/j.hrthm.2020.09.007. Epub 2020 Sep 12. Heart Rhythm. 2021. PMID: 32931925 Free PMC article.
-
Sudden cardiac death and genetic ion channelopathies: long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation.Circulation. 2012 Apr 24;125(16):2027-34. doi: 10.1161/CIRCULATIONAHA.111.055947. Circulation. 2012. PMID: 22529064 No abstract available.
-
Brugada Syndrome: Different Experimental Models and the Role of Human Cardiomyocytes From Induced Pluripotent Stem Cells.J Am Heart Assoc. 2022 Apr 5;11(7):e024410. doi: 10.1161/JAHA.121.024410. Epub 2022 Mar 24. J Am Heart Assoc. 2022. PMID: 35322667 Free PMC article. Review.
-
Sudden death and ion channel disease: pathophysiology and implications for management.Heart. 2011 Sep;97(17):1365-72. doi: 10.1136/hrt.2011.223883. Epub 2011 Jun 16. Heart. 2011. PMID: 21685181 Review.
References
-
- Zhao Z., Li X., El-Battrawy I., Lan H., Zhong R., Xu Q., Huang M., Liao Z., Lang S., Zimmermann W.-H., et al. Drug Testing in Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes from a Patient With Short QT Syndrome Type 1. Clin. Pharmacol. Ther. 2019;106:642–651. doi: 10.1002/cpt.1449. - DOI - PubMed
-
- El-Battrawy I., Zhao Z., Lan H., Li X., Yücel G., Lang S., Sattler K., Schünemann J.-D., Zimmermann W.-H., Cyganek L., et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom. Precis. Med. 2018;11:e001893. doi: 10.1161/CIRCGEN.117.001893. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
