

Complement System and Adhesion Molecule Skirmishes in Fabry Disease: Insights into Pathogenesis and Disease Mechanisms
- PMID: 39596318
- PMCID: PMC11594573
- DOI: 10.3390/ijms252212252
Complement System and Adhesion Molecule Skirmishes in Fabry Disease: Insights into Pathogenesis and Disease Mechanisms
Abstract
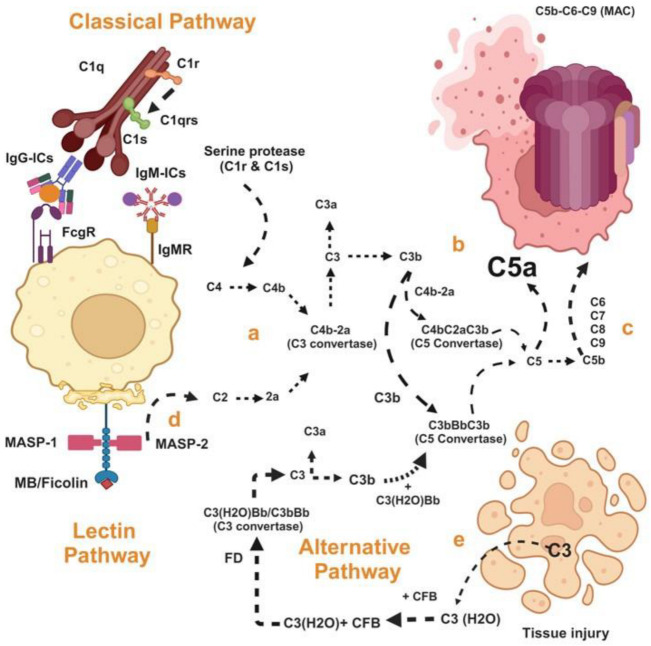
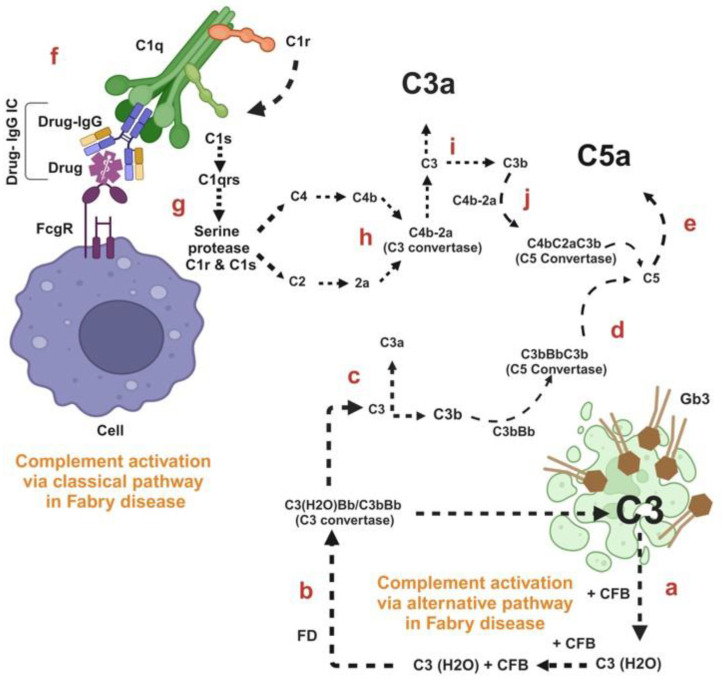
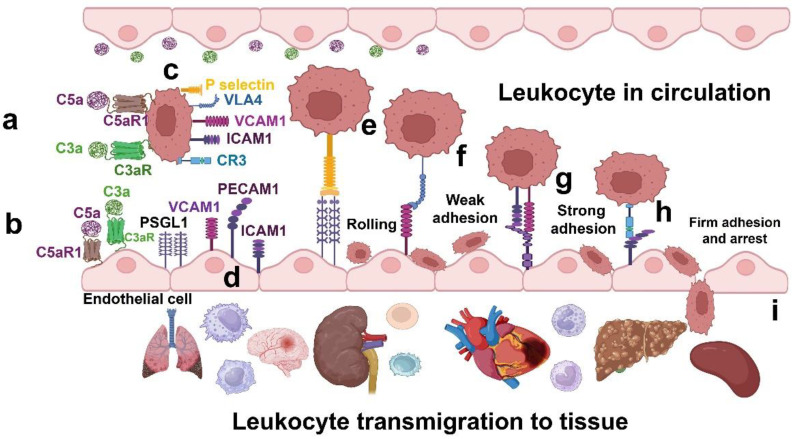
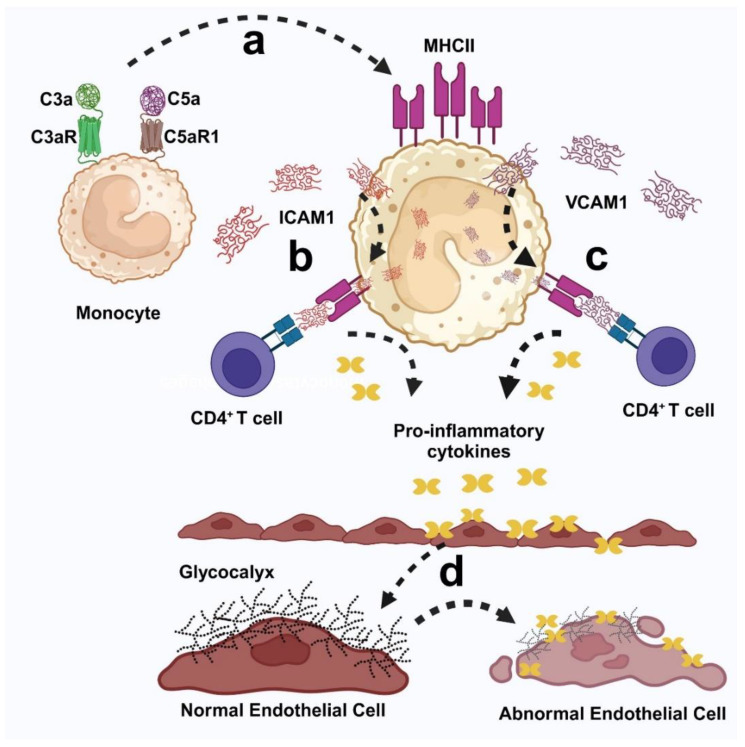
Fabry disease is a rare X-linked lysosomal storage disorder caused by mutations in the galactosidase alpha (GLA) gene, resulting in the accumulation of globotriaosylceramide (Gb3) and its deacetylated form, globotriaosylsphingosine (Lyso-Gb3) in various tissues and fluids throughout the body. This pathological accumulation triggers a cascade of processes involving immune dysregulation and complement system activation. Elevated levels of complement 3a (C3a), C5a, and their precursor C3 are observed in the plasma, serum, and tissues of patients with Fabry disease, correlating with significant endothelial cell abnormalities and vascular dysfunction. This review elucidates how the complement system, particularly through the activation of C3a and C5a, exacerbates disease pathology. The activation of these pathways leads to the upregulation of adhesion molecules, including vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), platelet and endothelial cell adhesion molecule 1 (PECAM1), and complement receptor 3 (CR3) on leukocytes and endothelial cells. This upregulation promotes the excessive recruitment of leukocytes, which in turn exacerbates disease pathology. Targeting complement components C3a, C5a, or their respective receptors, C3aR (C3a receptor) and C5aR1 (C5a receptor 1), could potentially reduce inflammation, mitigate tissue damage, and improve clinical outcomes for individuals with Fabry disease.
Keywords: cell adhesion molecules; complement-mediated injury; endothelial dysfunction; immune cell infiltration; inflammatory cascade.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Complement activation and cellular inflammation in Fabry disease patients despite enzyme replacement therapy.Front Immunol. 2024 Jan 18;15:1307558. doi: 10.3389/fimmu.2024.1307558. eCollection 2024. Front Immunol. 2024. PMID: 38304433 Free PMC article.
-
Pathogenesis and Molecular Mechanisms of Anderson-Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies.Int J Mol Sci. 2021 Sep 18;22(18):10088. doi: 10.3390/ijms221810088. Int J Mol Sci. 2021. PMID: 34576250 Free PMC article. Review.
-
Cardiac manifestations of Fabry disease in G3Stg/GlaKO and GlaKO mouse models-Translation to Fabry disease patients.PLoS One. 2024 May 31;19(5):e0304415. doi: 10.1371/journal.pone.0304415. eCollection 2024. PLoS One. 2024. PMID: 38820517 Free PMC article.
-
Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease.PLoS One. 2012;7(4):e36373. doi: 10.1371/journal.pone.0036373. Epub 2012 Apr 30. PLoS One. 2012. PMID: 22558451 Free PMC article.
-
Inflammation, Oxidative Stress, and Endothelial Dysfunction in the Pathogenesis of Vascular Damage: Unraveling Novel Cardiovascular Risk Factors in Fabry Disease.Int J Mol Sci. 2024 Jul 29;25(15):8273. doi: 10.3390/ijms25158273. Int J Mol Sci. 2024. PMID: 39125842 Free PMC article. Review.
Cited by
-
Molecular Diplomacy of Lipids in the War of Immunity: Bridging Rare and Common Disease Mechanisms.Int J Mol Sci. 2025 Jun 10;26(12):5568. doi: 10.3390/ijms26125568. Int J Mol Sci. 2025. PMID: 40565032 Free PMC article.
References
-
- Burlina A.B., Polo G., Salviati L., Duro G., Zizzo C., Dardis A., Bembi B., Cazzorla C., Rubert L., Zordan R., et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018;41:209–219. doi: 10.1007/s10545-017-0098-3. - DOI - PubMed
-
- Eng C.M., Niehaus D.J., Enriquez A.L., Burgert T.S., Ludman M.D., Desnick R. Fabry disease: Twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the α-galactosidase A gene. Hum. Mol. Genet. 1994;3:1795–1799. doi: 10.1093/hmg/3.10.1795. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
