

Phenotypic and Genetic Spectrum in 309 Consecutive Pediatric Patients with Inherited Retinal Disease
- PMID: 39596324
- PMCID: PMC11595089
- DOI: 10.3390/ijms252212259
Phenotypic and Genetic Spectrum in 309 Consecutive Pediatric Patients with Inherited Retinal Disease
Abstract
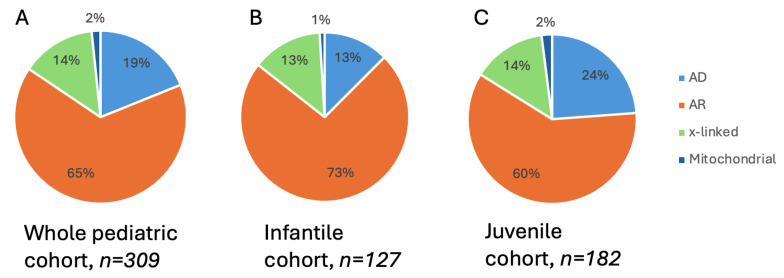
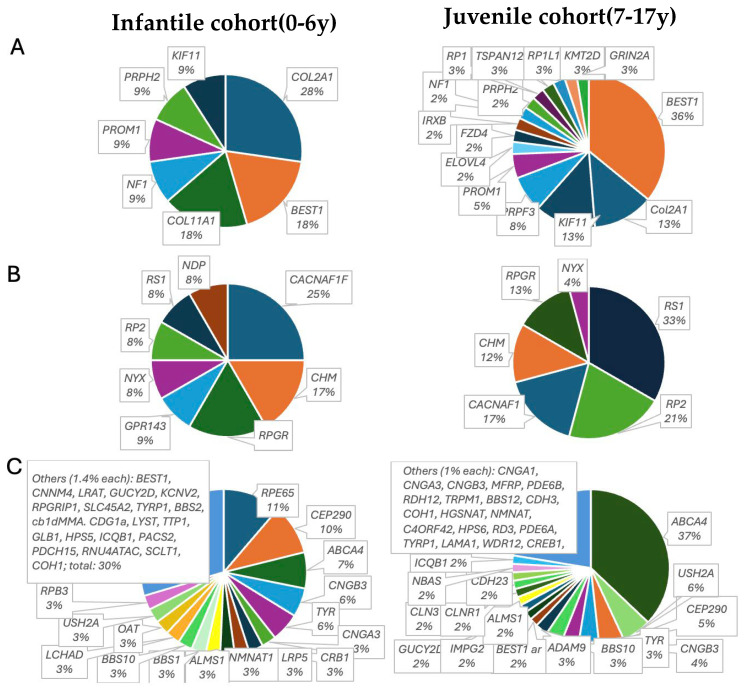
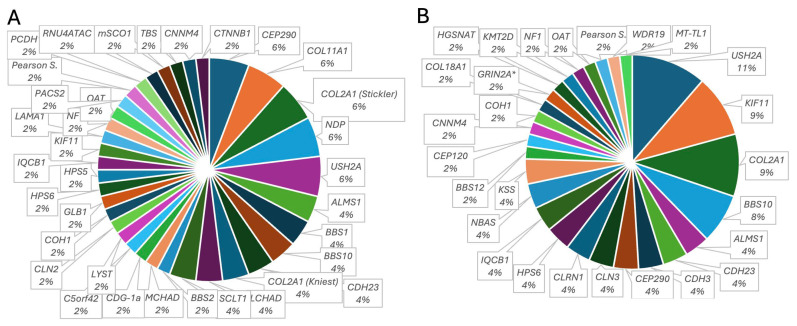
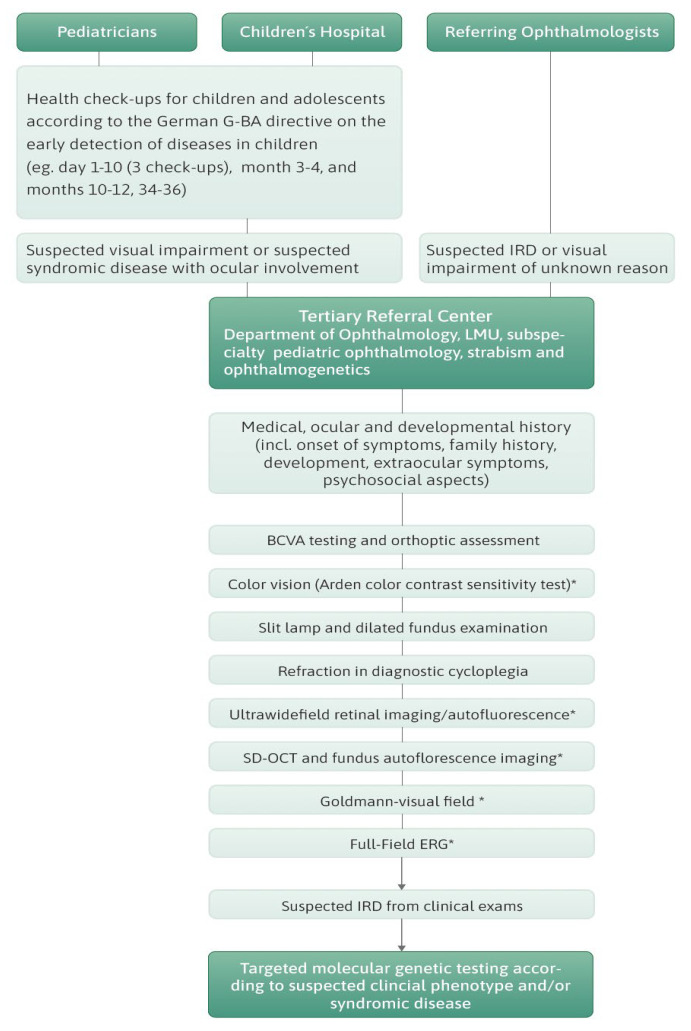
Inherited retinal dystrophies (IRDs) are a common cause of blindness or severe visual impairment in children and may occur with or without systemic associations. The aim of the present study is to describe the phenotypic and genotypic spectrum of IRDs in a pediatric patient cohort in Retrospective single-center cross-sectional analysis. Presenting symptoms, clinical phenotype, and molecular genetic diagnosis were assessed in 309 pediatric patients with suspected IRD. Patients were grouped by age at genetic diagnosis (preschool: 0-6 years, n = 127; schoolchildren: 7-17 years, n = 182). Preschool children most frequently presented with nystagmus (34.5% isolated, 16.4% syndromic), no visual interest (20.9%; 14.5%), or nyctalopia (22.4%; 3.6%; p < 0.05); schoolchildren most frequently presented with declining visual acuity (31% isolated, 21.1% syndromic), nyctalopia (10.6%; 13.5%), or high myopia (5.3%; 13.2%). Pathogenic variants were identified in 96 different genes (n = 69 preschool, n = 73 schoolchildren). In the preschool group, 57.4% had isolated and 42.6% had syndromic IRDs, compared to 70.9% and 29.1% in schoolchildren. In the preschool group, 32.4% of the isolated IRDs were related to forms of Leber's congenital amaurosis (most frequent were RPE65 (11%) and CEP290 (8.2%)), 31.5% were related to stationary IRDs, 15.1% were related to macular dystrophies (ABCA4, BEST1, PRPH2, PROM1), and 8.2% to rod-cone dystrophies (RPGR, RPB3, RP2, PDE6A). All rod-cone dystrophies (RCDs) were subjectively asymptomatic at the time of genetic diagnosis. At schoolage, 41% were attributed to cone-dominated disease (34% ABCA4), 10.3% to BEST1, and 10.3% to RCDs (RP2, PRPF3, RPGR; IMPG2, PDE6B, CNGA1, MFRP, RP1). Ciliopathies were the most common syndromic IRDs (preschool 37%; schoolchildren 45.1%), with variants in USH2A, CEP290 (5.6% each), CDH23, BBS1, and BBS10 (3.7% each) being the most frequent in preschoolers, and USH2A (11.7%), BBS10 (7.8%), CEP290, CDHR23, CLRN1, and ICQB1 (3.9% each) being the most frequent in syndromic schoolkids. Vitreoretinal syndromic IRDs accounted for 29.6% (preschool: COL2A1, COL11A1, NDP (5.6% each)) and 23.5% (schoolage: COL2A1, KIF11 (9.8% each)), metabolic IRDs for 9.4% (OAT, HADHA, MMACHD, PMM2) and 3.9% (OAT, HADHA), mitochondriopathies for 3.7% and 7.8%, and syndromic albinism accounted for 5.6% and 3.9%, respectively. In conclusion we show here that the genotypic spectrum of IRDs and its quantitative distribution not only differs between children and adults but also between children of different age groups, with an almost equal proportion of syndromic and non-syndromic IRDs in early childhood. Ophthalmic screening visits at the preschool and school ages may aid even presymptomatic diagnosis and treatment of potential sight and life-threatening systemic sequelae.
Keywords: inherited retinal dystrophy; pediatric patients; syndromic retinal dystrophies.
Conflict of interest statement
Claudia Priglinger received speaker fees from Novartis Pharma GmbH and Chiesi, Hanno Jörn Bolz was employed at Bioscientia Institute of Medical Diagnostics GmbH. Teresa M. Neuhann is partner at MGZ—Medizinisch Genetisches Zentrum, and medical advisor for Janssen and Chiesi; Maximilian Gerhardt received speaker fees from Novartis Pharma GmbH, Bayer Pharma, and Janssen. Siegfried Priglinger obtained advisory board honoraria from Novartis Pharma GmbH, Pharm Allergan, Alcon, Zeiss, BVI, B&L, Roche, and AbbVie and is medical consultant for Zeiss, BVI, B&L, Roche and AbbVie. Markus Schaumberger, Guenther Rudolph and Yasemin Mehraein have no conflicts of interest to declare. The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Weisschuh N., Obermaier C.D., Battke F., Bernd A., Kuehlewein L., Nasser F., Zobor D., Zrenner E., Weber E., Wissinger B., et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020;41:1514–1527. doi: 10.1002/humu.24064. - DOI - PubMed
-
- De Silva S.R., Arno G., Robson A.G., Fakin A., Pontikos N., Mohamed M.D., Bird A.C., Moore A.T., Michaelides M., Webster A.R., et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021;82:100898. doi: 10.1016/j.preteyeres.2020.100898. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
