

Investigation of Polymorphisms Induced by the Solo Long Terminal Repeats (Solo-LTRs) in Porcine Endogenous Retroviruses (ERVs)
- PMID: 39599915
- PMCID: PMC11598996
- DOI: 10.3390/v16111801
Investigation of Polymorphisms Induced by the Solo Long Terminal Repeats (Solo-LTRs) in Porcine Endogenous Retroviruses (ERVs)
Abstract
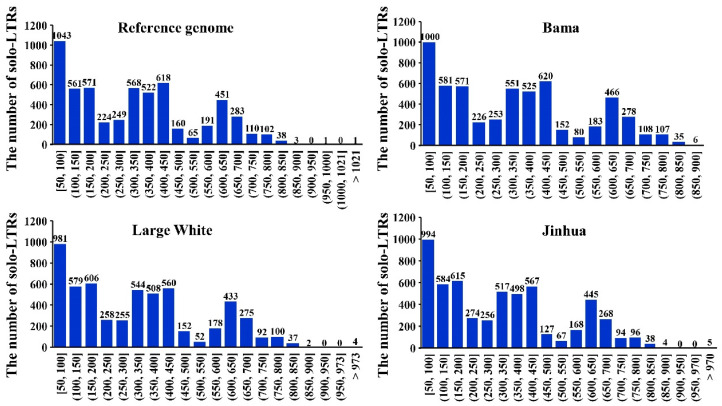
Homologous recombination events take place between the 5' and 3' long terminal repeats (LTRs) of ERVs, resulting in the generation of solo-LTR, which can cause solo-LTR-associated polymorphism across different genomes. In the current study, specific criteria were established for the filtration of solo-LTRs, resulting in an average of 5630 solo-LTRs being identified in 21 genomes. Subsequently, a protocol was developed for detecting solo-LTR polymorphisms in the pig genomes, resulting in the discovery of 927 predicted solo-LTR polymorphic sites. Following verification and filtration processes, 603 highly reliable solo-LTR polymorphic sites were retained, involving 446 solo-LTR presence sites (solo-LTR+) and 157 solo-LTR absence sites (solo-LTR-) relative to the reference genome. Intersection analysis with gene/functional regions revealed that 248 solo-LTR- sites and 23 solo-LTR+ sites overlapped with genes or were in the vicinity of genes or functional regions, impacting a diverse range of gene structures. Moreover, through the utilization of 156 solo-LTR polymorphic sites for population genetic analysis, it was observed that these solo-LTR loci effectively clustered various breeds together, aligning with expectations and underscoring their practical utility. This study successfully established a methodology for detecting solo-LTR polymorphic sites. By applying these methods, a total of 603 high-reliability solo-LTR polymorphic sites were pinpointed, with nearly half of them being linked to genes or functional regions.
Keywords: genomic variation; polymorphic sites; population genetics; solo-LTR.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



References
Publication types
MeSH terms
Grants and funding
- JBGS(2021)028)/the Revitalization of Seed Industry (JBGS) in Jiangsu province
- IJRLD-KF202209/the Open Project Program of International Joint Research Laboratory in Universities of Jiangsu Province of China for Domestic Animal Germplasm Resources and Genetic Improvement
- NA/the High-end Talent Support Program of Yangzhou University to Chengyi Song
LinkOut - more resources
Full Text Sources
