

Constitutive opening of the Kv7.2 pore activation gate causes KCNQ2-developmental encephalopathy
- PMID: 39602259
- PMCID: PMC11626135
- DOI: 10.1073/pnas.2412388121
Constitutive opening of the Kv7.2 pore activation gate causes KCNQ2-developmental encephalopathy
Abstract
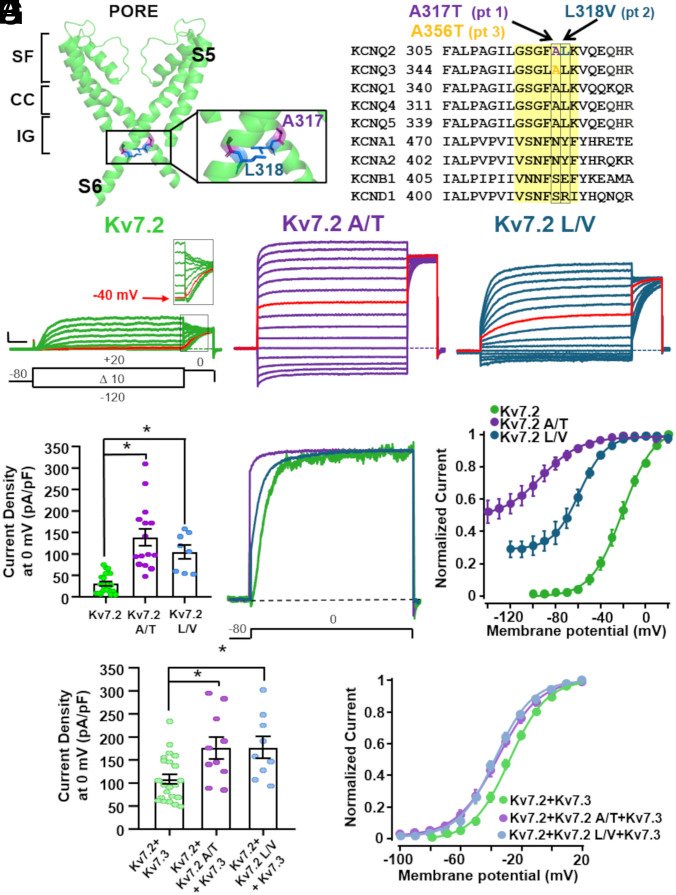
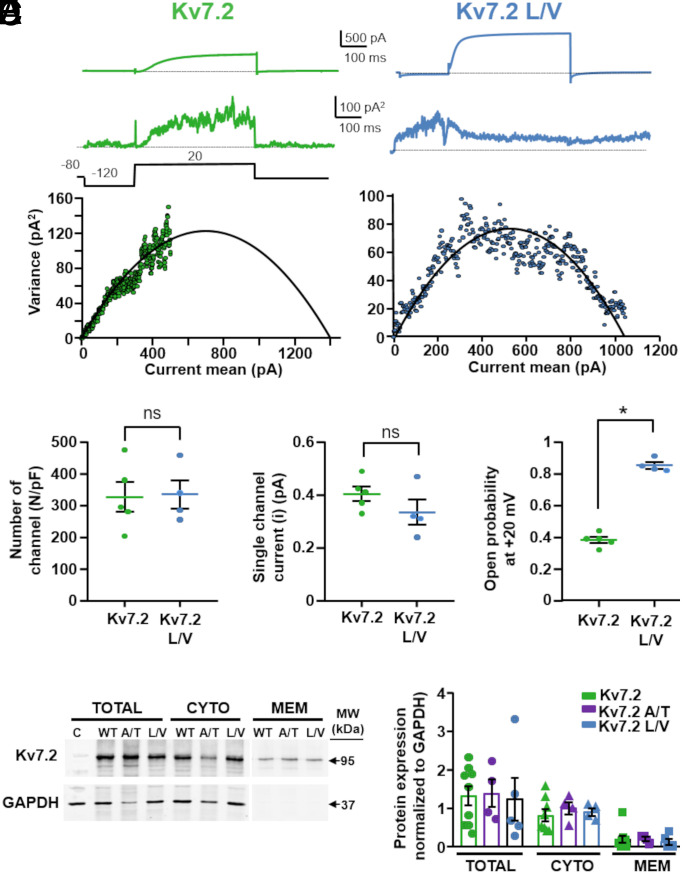
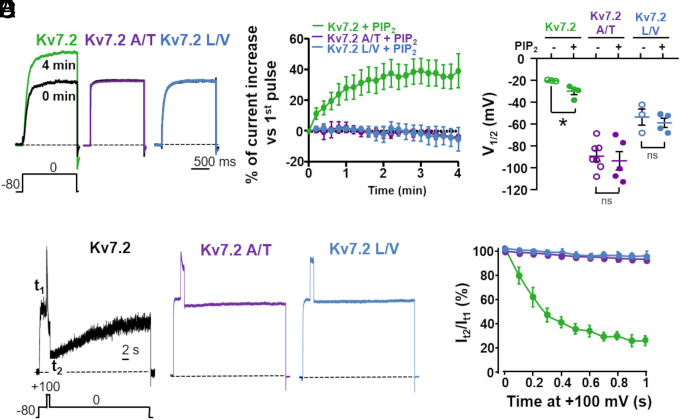
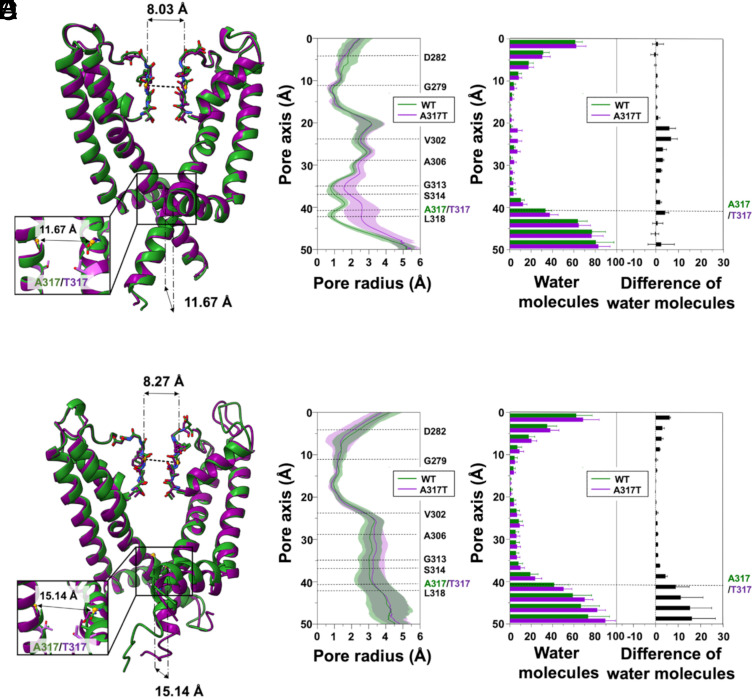
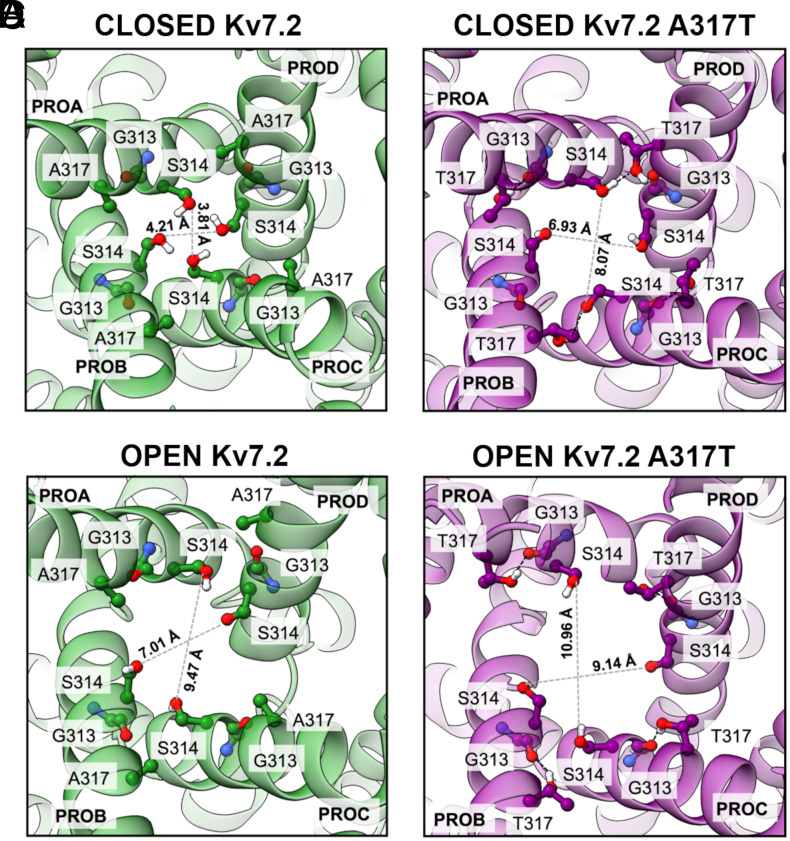
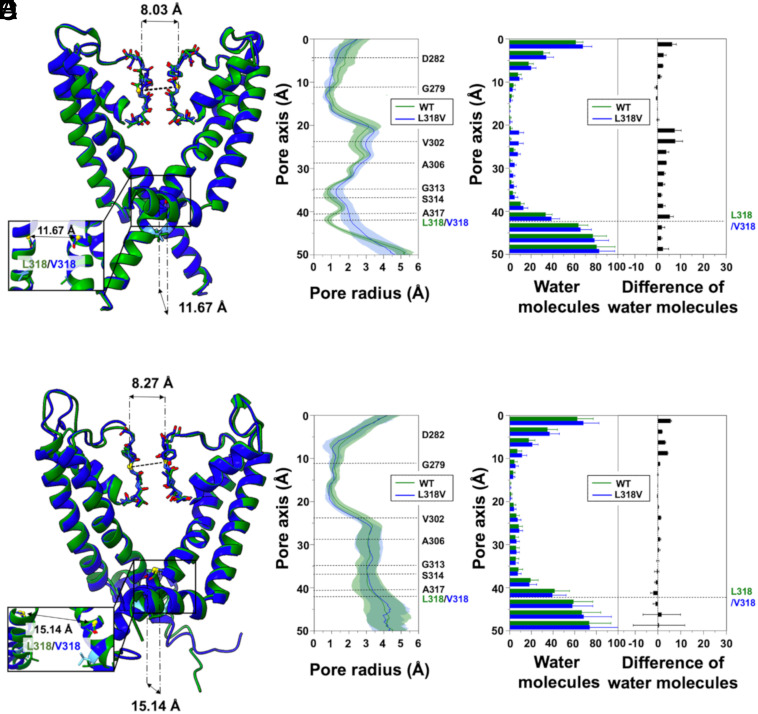
Pathogenic variants in KCNQ2 encoding Kv7.2 voltage-gated potassium channel subunits cause developmental encephalopathies (KCNQ2-encephalopathies), both with and without epilepsy. We herein describe the clinical, in vitro, and in silico features of two encephalopathy-causing variants (A317T, L318V) in Kv7.2 affecting two consecutive residues in the S6 activation gate that undergoes large structural rearrangements during pore opening; the disease-causing A356T variant in KCNQ3, paralogous to the A317T variant in KCNQ2, was also investigated. Currents through KCNQ2 mutant channels displayed increased density, hyperpolarizing shifts in activation gating, faster activation and slower deactivation kinetics, and resistance to changes in the cellular concentrations of phosphatidylinositol 4,5-bisphosphate (PIP2), a critical regulator of Kv7 channel function; all these features are consistent with a strong gain-of-function effect. An increase in the probability of single-channel opening, with no change in membrane abundance or single-channel conductance, was responsible for the observed gain-of-function effects. All-atom molecular dynamics simulations revealed that the mutations widened the inner pore gate and stabilized a constitutively open channel configuration in the closed state, with minimal effects on the open conformation. Thus, mutation-induced stabilization of the inner pore gate open configuration is a molecular pathogenetic mechanism for KCNQ2-related encephalopathies.
Keywords: Genotype–phenotype correlations; Molecular dynamics; channel gating; developmental and epileptic encephalopathies; potassium channels.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures
References
-
- Weckhuysen S., et al. , KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25 (2012). - PubMed
-
- Nappi P., et al. , Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflugers Arch. 472, 881–898 (2020). - PubMed
-
- Miceli F., Soldovieri M. V., Weckhuysen S., Cooper E., Taglialatela M., “KCNQ2-Related Disorders” in GeneReviews®, Adam M. P., et al. , Eds. (University of Washington, Seattle, 1993). - PubMed
-
- Miceli F., Soldovieri M. V., Weckhuysen S., Cooper E. C., Taglialatela M., “KCNQ3-Related Disorders” in GeneReviews®, Adam M. P., et al. , Eds. (University of Washington, Seattle, 1993). - PubMed
-
- Zuberi S. M., et al. , ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE task force on nosology and definitions. Epilepsia 63, 1349–1397 (2022). - PubMed
MeSH terms
Substances
Grants and funding
- RF-2019-12370491/Ministero della Salute (Italy Ministry of Health)
- 1861419 N G041821N/Fonds Wetenschappelijk Onderzoek (FWO)
- PRIN 2020XBFEMS/Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR)
- PNRR-MR1-2022-12376528/Ministero della Salute (Italy Ministry of Health)
- TREATKCNQ/European Joint Programme on Rare Disease JTC 2020
LinkOut - more resources
Full Text Sources
