

Transcriptomics of interstitial lung disease: a systematic review and meta-analysis
- PMID: 39603671
- PMCID: PMC12138033
- DOI: 10.1183/13993003.01070-2024
Transcriptomics of interstitial lung disease: a systematic review and meta-analysis
Abstract
Objective: Gene expression (transcriptomics) studies have revealed potential mechanisms of interstitial lung disease, yet sample sizes of studies are often limited and between-subtype comparisons are scarce. The aim of this study was to identify and validate consensus transcriptomic signatures of interstitial lung disease subtypes.
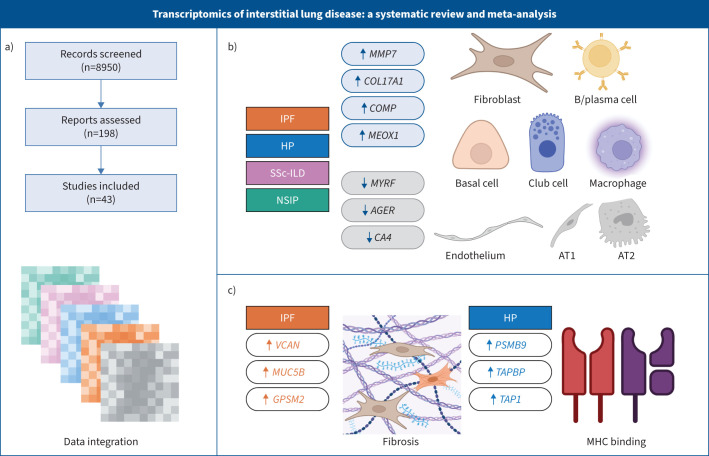
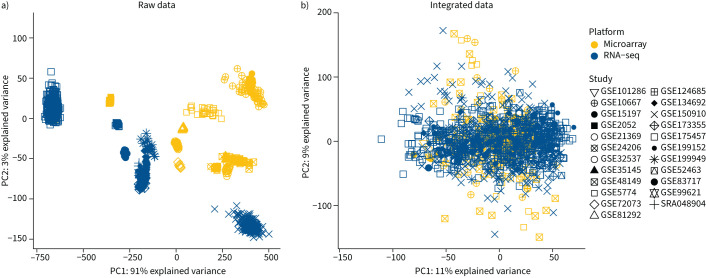
Methods: We performed a systematic review and meta-analysis of fibrotic interstitial lung disease transcriptomics studies using an individual participant data approach. We included studies examining bulk transcriptomics of human adult interstitial lung disease samples and excluded those focusing on individual cell populations. Patient-level data and expression matrices were extracted from 43 studies and integrated using a multivariable integrative algorithm to develop interstitial lung disease classification models.
Results: Using 1459 samples from 24 studies, we identified transcriptomic signatures for idiopathic pulmonary fibrosis, hypersensitivity pneumonitis, idiopathic nonspecific interstitial pneumonia and systemic sclerosis-associated interstitial lung disease against control samples, which were validated on 308 samples from eight studies (idiopathic pulmonary fibrosis area under receiver operating curve (AUC) 0.99, 95% CI 0.99-1.00; hypersensitivity pneumonitis AUC 0.91, 95% CI 0.84-0.99; nonspecific interstitial pneumonia AUC 0.94, 95% CI 0.88-0.99; systemic sclerosis-associated interstitial lung disease AUC 0.98, 95% CI 0.93-1.00). Significantly, meta-analysis allowed us to identify, for the first time, robust lung transcriptomics signatures to discriminate idiopathic pulmonary fibrosis (AUC 0.71, 95% CI 0.63-0.79) and hypersensitivity pneumonitis (AUC 0.76, 95% CI 0.63-0.89) from other fibrotic interstitial lung disease, and unsupervised learning algorithms identified putative molecular endotypes of interstitial lung disease associated with decreased forced vital capacity and diffusing capacity of the lungs for carbon monoxide % predicted. Transcriptomics signatures were reflective of both cell-specific and disease-specific changes in gene expression.
Conclusion: We present the first systematic review and largest meta-analysis of fibrotic interstitial lung disease transcriptomics to date, identifying reproducible transcriptomic signatures with clinical relevance.
Copyright ©The authors 2025.
Conflict of interest statement
Conflict of interest: S.A. Guler reports grants from Boehringer Ingelheim, Roche, MSD and Janssen; payment or honoraria for lectures, presentations, manuscript writing or educational events and support for attending meetings from Boehringer Ingelheim; and participation on a data safety monitoring board or advisory board with Boehringer Ingelheim, MSD and Janssen. C.J. Ryerson reports grants from Boehringer Ingelheim; consulting fees from Boehringer Ingelheim, Pliant Therapeutics, AstraZeneca, Trevi Therapeutics and Veracyte; payment or honoraria for lectures, presentations, manuscript writing or educational events from Hoffmann-La Roche, Boehringer Ingelheim and Cipla; payment for expert testimony from Boehringer Ingelheim; and support for attending meetings from Cipla and Boehringer Ingelheim. The remaining authors have no potential conflicts of interest to disclose.
Figures





Comment in
-
Climbing the hierarchy of evidence in interstitial lung disease transcriptomics.Eur Respir J. 2025 Jun 5;65(6):2402471. doi: 10.1183/13993003.02471-2024. Print 2025 Jun. Eur Respir J. 2025. PMID: 40473299 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical