

This is a preprint.
Extensive binding of uncharacterized human transcription factors to genomic dark matter
- PMID: 39605320
- PMCID: PMC11601254
- DOI: 10.1101/2024.11.11.622123
Extensive binding of uncharacterized human transcription factors to genomic dark matter
Abstract
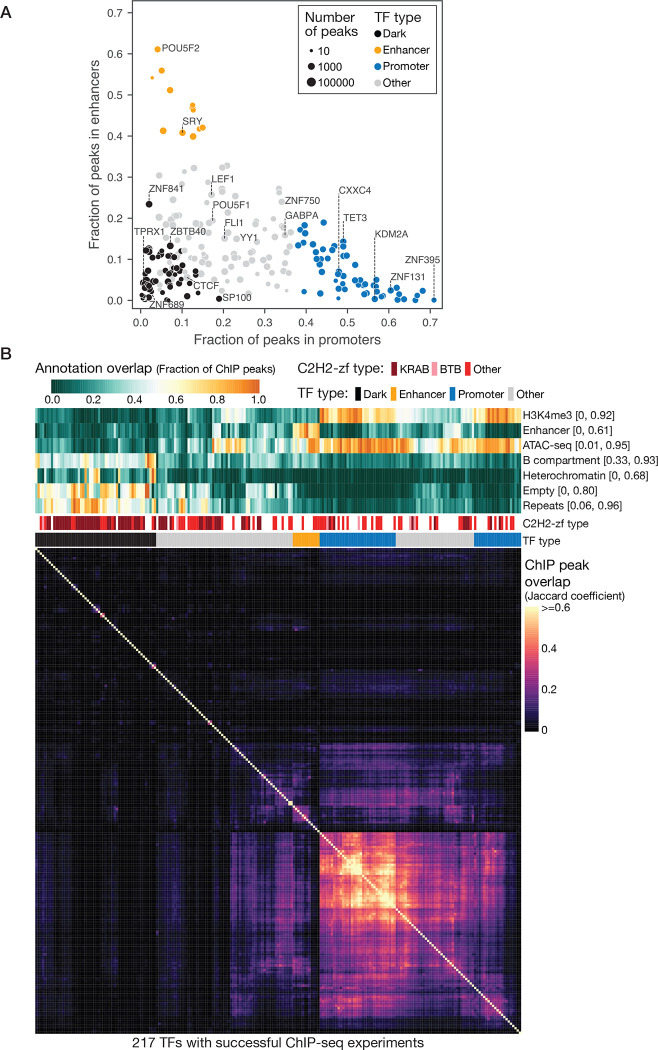
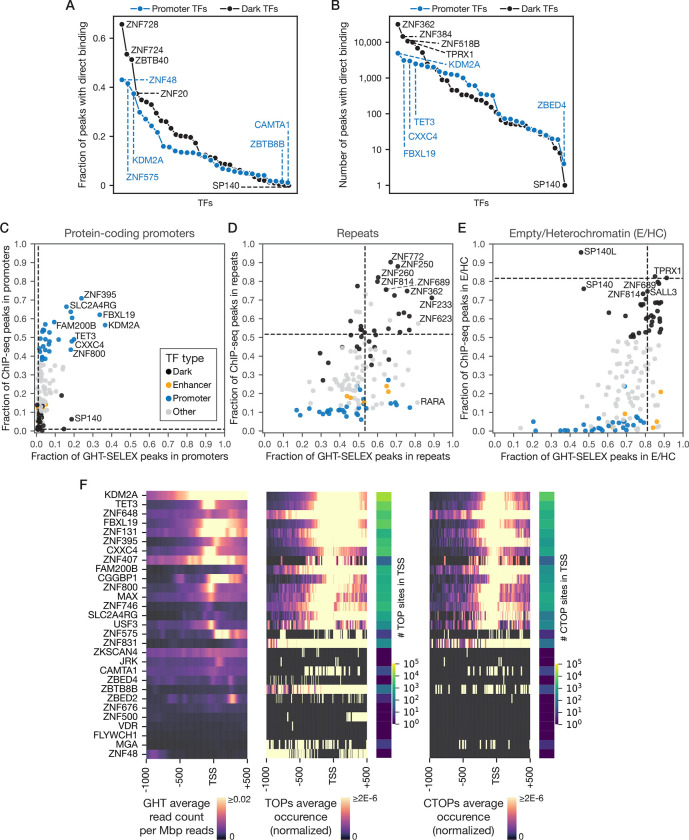
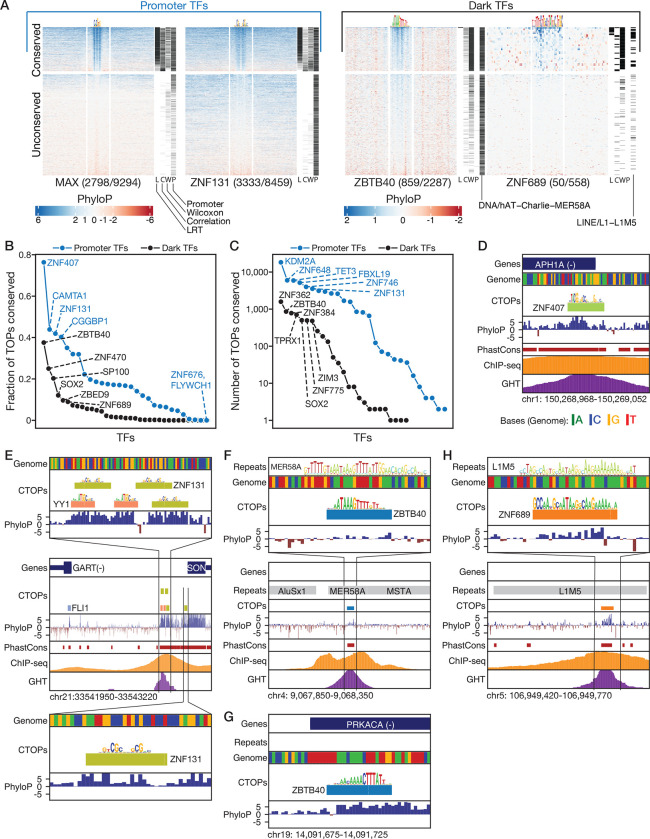
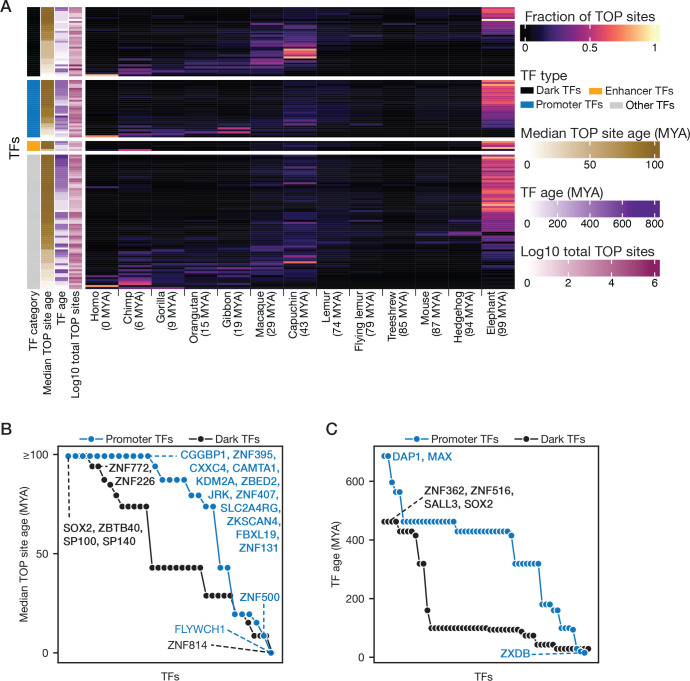
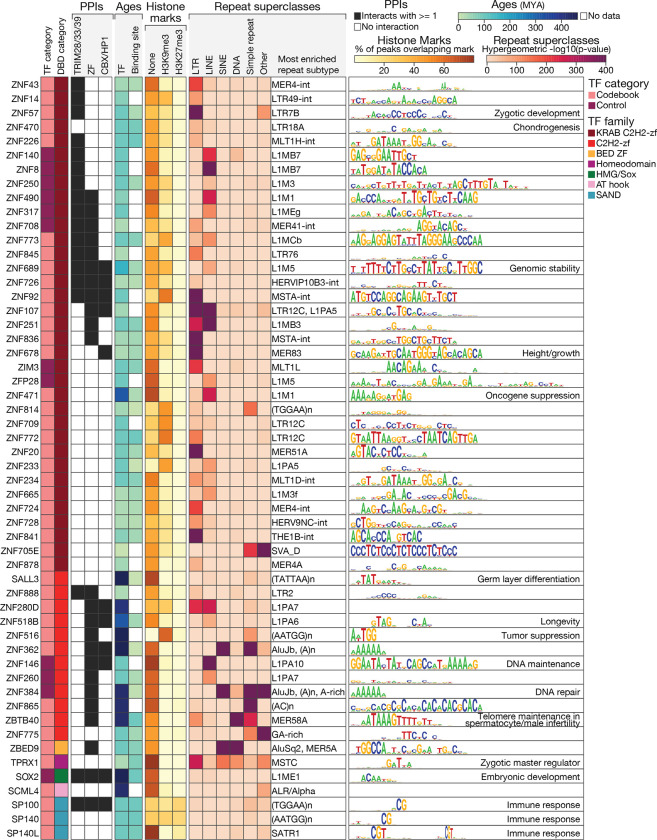
Most of the human genome is thought to be non-functional, and includes large segments often referred to as "dark matter" DNA. The genome also encodes hundreds of putative and poorly characterized transcription factors (TFs). We determined genomic binding locations of 166 uncharacterized human TFs in living cells. Nearly half of them associated strongly with known regulatory regions such as promoters and enhancers, often at conserved motif matches and co-localizing with each other. Surprisingly, the other half often associated with genomic dark matter, at largely unique sites, via intrinsic sequence recognition. Dozens of these, which we term "Dark TFs", mainly bind within regions of closed chromatin. Dark TF binding sites are enriched for transposable elements, and are rarely under purifying selection. Some Dark TFs are KZNFs, which contain the repressive KRAB domain, but many are not: the Dark TFs also include known or potential pioneer TFs. Compiled literature information supports that the Dark TFs exert diverse functions ranging from early development to tumor suppression. Thus, our results sheds light on a large fraction of previously uncharacterized human TFs and their unappreciated activities within the dark matter genome.
Keywords: C2H2; ChIP-seq; Codebook; GHT-SELEX; Gene regulation; KRAB zinc finger protein; PWM; SELEX; Transcription factor (TF).
Figures


References
-
- Lambert S.A. et al. The Human Transcription Factors. Cell 175, 598–599 (2018). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous