

This is a preprint.
In vivo Treatment of a Severe Vascular Disease via a Bespoke CRISPR-Cas9 Base Editor
- PMID: 39605323
- PMCID: PMC11601241
- DOI: 10.1101/2024.11.11.621817
In vivo Treatment of a Severe Vascular Disease via a Bespoke CRISPR-Cas9 Base Editor
Update in
-
Treatment of a severe vascular disease using a bespoke CRISPR-Cas9 base editor in mice.Nat Biomed Eng. 2025 Sep 11. doi: 10.1038/s41551-025-01499-1. Online ahead of print. Nat Biomed Eng. 2025. PMID: 40935887
Abstract
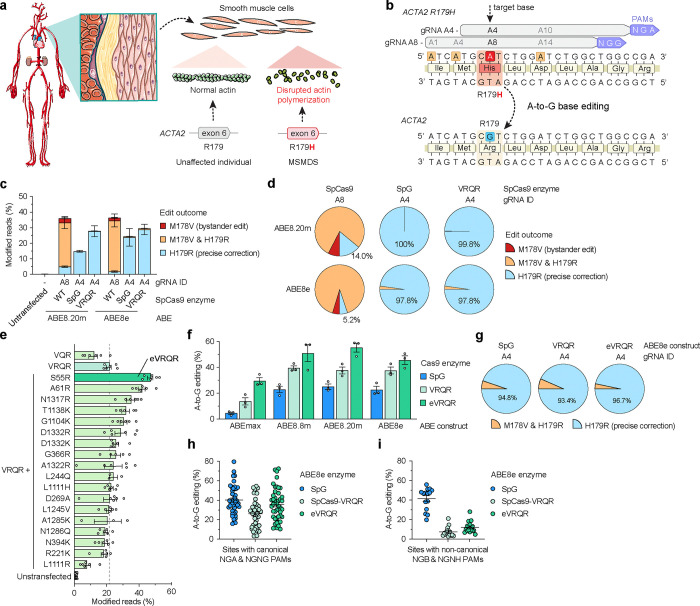
Genetic vascular disorders are prevalent diseases that have diverse etiologies and few treatment options. Pathogenic missense mutations in the alpha actin isotype 2 gene (ACTA2) primarily affect smooth muscle cell (SMC) function and cause multisystemic smooth muscle dysfunction syndrome (MSMDS), a genetic vasculopathy that is associated with stroke, aortic dissection, and death in childhood. Here, we explored genome editing to correct the most common MSMDS-causative mutation ACTA2 R179H. In a first-in-kind approach, we performed mutation-specific protein engineering to develop a bespoke CRISPR-Cas9 enzyme with enhanced on-target activity against the R179H sequence. To directly correct the R179H mutation, we screened dozens of configurations of base editors (comprised of Cas9 enzymes, deaminases, and gRNAs) to develop a highly precise corrective A-to-G edit with minimal deleterious bystander editing that is otherwise prevalent when using wild-type SpCas9 base editors. We then created a murine model of MSMDS that exhibits phenotypes consistent with human patients, including vasculopathy and premature death, to explore the in vivo therapeutic potential of this base editing strategy. Delivery of the customized base editor via an engineered SMC-tropic adeno-associated virus (AAV-PR) vector substantially prolonged survival and rescued systemic phenotypes across the lifespan of MSMDS mice, including in the vasculature, aorta, and brain. Together, our optimization of a customized base editor highlights how bespoke CRISPR-Cas enzymes can enhance on-target correction while minimizing bystander edits, culminating in a precise editing approach that may enable a long-lasting treatment for patients with MSMDS.
Keywords: ACTA2; CRISPR-Cas; MDMS; MSMDS; aortic aneurysm; base editing; bystander editing; genome editing; moyamoya disease; pediatric disease; vasculopathies.
Conflict of interest statement
Competing interests C.L.C., R.M., C.A.M., D.Y.C., B.P.K., M.E.L., and P.L.M. are inventors on a patent application filed by Mass General Brigham (MGB) that describes the development of genome editing technologies to treat MSMDS. C.R.R.A, R.A.S., J.F.dS., and B.P.K. are inventors on additional patents or patent applications filed by MGB that describe genome engineering technologies. S.Q.T. is an inventor on a patent covering CHANGE-seq. S.Q.T. is a member of the scientific advisory board of Prime Medicine and Ensoma. R.M., D.Y.C., C.A.M., B.P.K., M.E.L., and P.M.M received sponsored research support from Angea Biotherapeutics, a company developing gene therapies for vasculopathies. R.M. receives research funding from Amgen and serves as a consultant for Pharmacosmos, Myokardia/BMS, Renovacor, Epizon Pharma, and Third Pole and performs speaker bureaus through Vox Media, all of which are unrelated to the current work. C.A.M. has financial interests in Chameleon Biosciences, Skylark Bio, and Sphere Gene Therapeutics, companies developing Adeno Associated Virus (AAV) vector technologies for gene therapy applications. C.A.M. performs paid consulting work for all three companies. C.A.M.’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. B.P.K. is a consultant for EcoR1 capital, Novartis Venture Fund, and Jumble Therapeutics, and is on the scientific advisory boards of Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. B.P.K.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. The other authors declare no competing interests.
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous