

This is a preprint.
Leveraging chromatin packing domains to target chemoevasion in vivo
- PMID: 39605341
- PMCID: PMC11601449
- DOI: 10.1101/2024.11.14.623612
Leveraging chromatin packing domains to target chemoevasion in vivo
Update in
-
Leveraging chromatin packing domains to target chemoevasion in vivo.Proc Natl Acad Sci U S A. 2025 Jul 29;122(30):e2425319122. doi: 10.1073/pnas.2425319122. Epub 2025 Jul 22. Proc Natl Acad Sci U S A. 2025. PMID: 40694328 Free PMC article.
Abstract
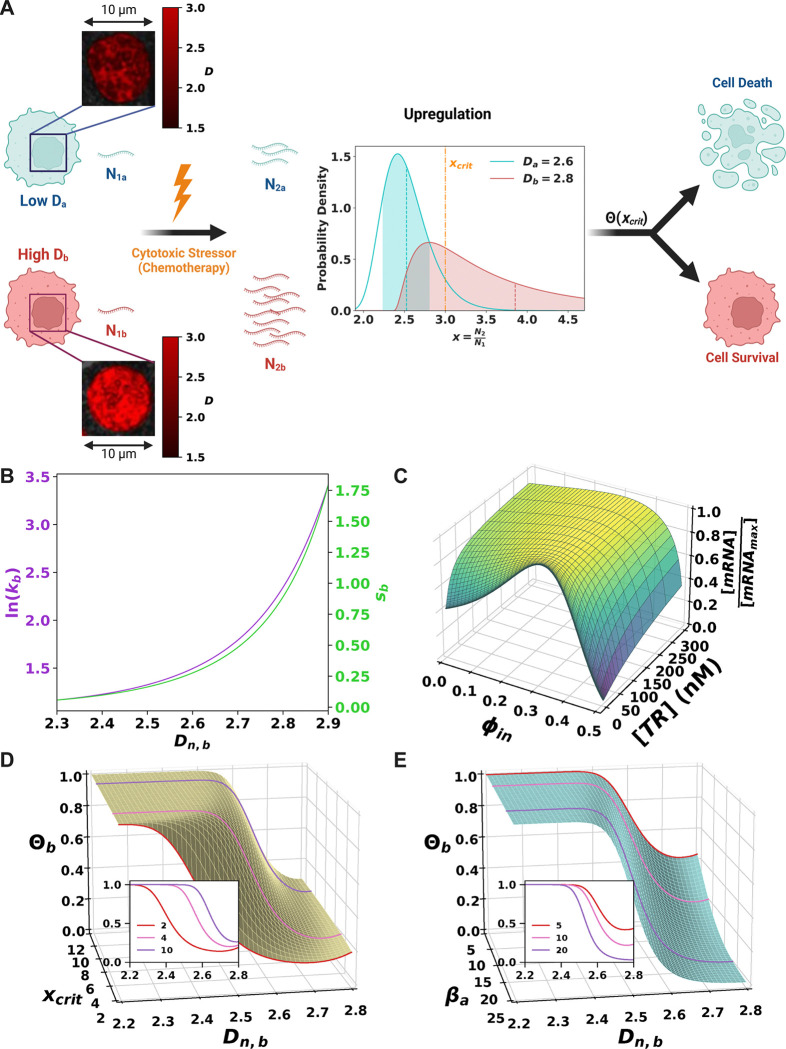
Cancer cells exhibit a remarkable resilience to cytotoxic stress, often adapting through transcriptional changes linked to alterations in chromatin structure. In several types of cancer, these adaptations involve epigenetic modifications and restructuring of topologically associating domains (TADs). However, the underlying principles by which chromatin architecture facilitates such adaptability across different cancers remain poorly understood. To investigate the role of chromatin in this process, we developed a physics-based mechanistic model that connects chromatin organization to cell fate decisions, specifically survival following chemotherapy. Our model builds on the observation that chromatin forms packing domains, which influence transcriptional efficiency through macromolecular crowding. The model accurately predicts chemoevasion in vitro, suggesting that changes in packing domains affect the likelihood of survival. Consistent results across diverse cancer types indicate that the model captures fundamental principles of chromatin-mediated adaptation, independent of the specific cancer or chemotherapy mechanisms involved. Based on these insights, we hypothesized that compounds capable of modulating packing domains, termed Transcriptional Plasticity Regulators (TPRs), could prevent cellular adaptation to chemotherapy. Using live-cell chromatin imaging, we conducted a compound screen that identified several TPRs which synergistically enhanced chemotherapy-induced cell death. The most effective TPR significantly improved therapeutic outcomes in a patient-derived xenograft (PDX) model of ovarian cancer. These findings underscore the central role of chromatin in cellular adaptation to cytotoxic stress and present a novel framework for enhancing cancer therapies, with broad potential across multiple cancer types.
Keywords: Biophysics; Cancer; Chemotherapy; Chromatin; Plasticity.
Figures




References
-
- Aas Turid, Børresen Anne-Lise, Geisler Stephanie, Smith-Sørensen Birgitte, Johnsen Hilde, Varhaug Jan E., Akslen Lars A., and Lønning Per E.. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nature Medicine, 2(7): 811–814, July 1996. ISSN 1546–170X. doi: 10.1038/nm0796-811. Number: 7 Publisher: Nature Publishing Group. - DOI - PubMed
-
- Lièvre Astrid, Bachet Jean-Baptiste, Le Corre Delphine, Boige Valérie, Landi Bruno, Emile Jean-François, Côté Jean-François, Tomasic Gorana, Penna Christophe, Ducreux Michel, Rougier Philippe, Penault-Llorca Frédérique, and Laurent-Puig Pierre. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Research, 66(8):3992–3995, April 2006. ISSN 0008–5472. doi: 10.1158/0008-5472. CAN-06-0191. - DOI - PubMed
-
- Sobhani Navid, Roviello Giandomenico, Corona Silvia P., Scaltriti Maurizio, Ianza Anna, Bortul Marina, Zanconati Fabrizio, and Generali Daniele. The prognostic value of PI3K mutational status in breast cancer: A meta-analysis. Journal of Cellular Biochemistry, 119(6):4287–4292, 2018. ISSN 1097–4644. doi: . _eprint: . - DOI - DOI - PMC - PubMed
-
- Martínez-Jiménez Francisco, Movasati Ali, Brunner Sascha Remy, Nguyen Luan, Priestley Peter, Cuppen Edwin, and Van Hoeck Arne. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature, pages 1–9, May 2023. ISSN 1476–4687. doi: 10.1038/s41586-023-06054-z. Publisher: Nature Publishing Group. - DOI - PMC - PubMed
-
- Gerstung Moritz, Jolly Clemency, Leshchiner Ignaty, Dentro Stefan C., Gonzalez Santiago, Rosebrock Daniel, Mitchell Thomas J., Rubanova Yulia, Anur Pavana, Yu Kaixian, Tarabichi Maxime, Deshwar Amit, Wintersinger Jeff, Kleinheinz Kortine, Vázquez-García Ignacio, Haase Kerstin, Jerman Lara, Sengupta Subhajit, Macintyre Geoff, Malikic Salem, Donmez Nilgun, Livitz Dimitri G., Cmero Marek, Demeulemeester Jonas, Schumacher Steven, Fan Yu, Yao Xiaotong, Lee Juhee, Schlesner Matthias, Boutros Paul C., Bowtell David D., Zhu Hongtu, Getz Gad, Imielinski Marcin, Beroukhim Rameen, Sahinalp S. Cenk, Ji Yuan, Peifer Martin, Markowetz Florian, Mustonen Ville, Yuan Ke, Wang Wenyi, Morris Quaid D., Spellman Paul T., Wedge David C., and Van Loo Peter. The evolutionary history of 2,658 cancers. Nature, 578(7793):122–128, February 2020. ISSN 1476–4687. doi: 10.1038/s41586-019-1907-7. Number: 7793 Publisher: Nature Publishing Group. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources