

This is a preprint.
Genotype-immunophenotype relationships in NPM1-mutant AML clonal evolution uncovered by single cell multiomic analysis
- PMID: 39605444
- PMCID: PMC11601460
- DOI: 10.1101/2024.11.11.623033
Genotype-immunophenotype relationships in NPM1-mutant AML clonal evolution uncovered by single cell multiomic analysis
Abstract
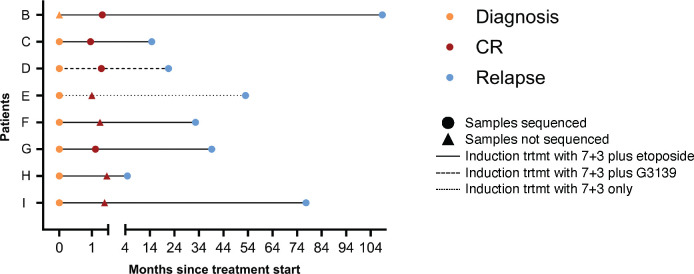
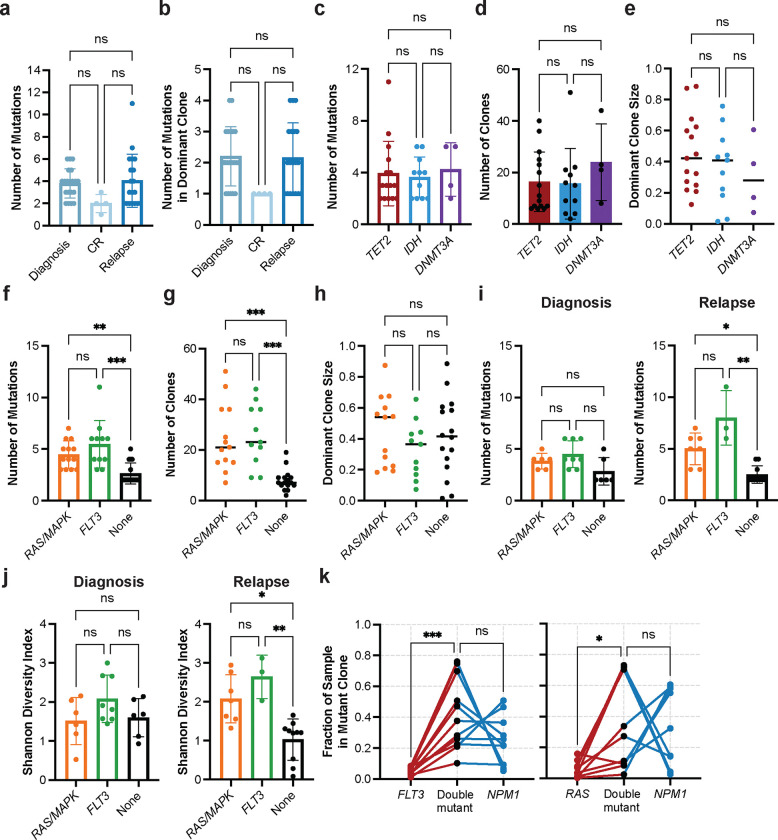
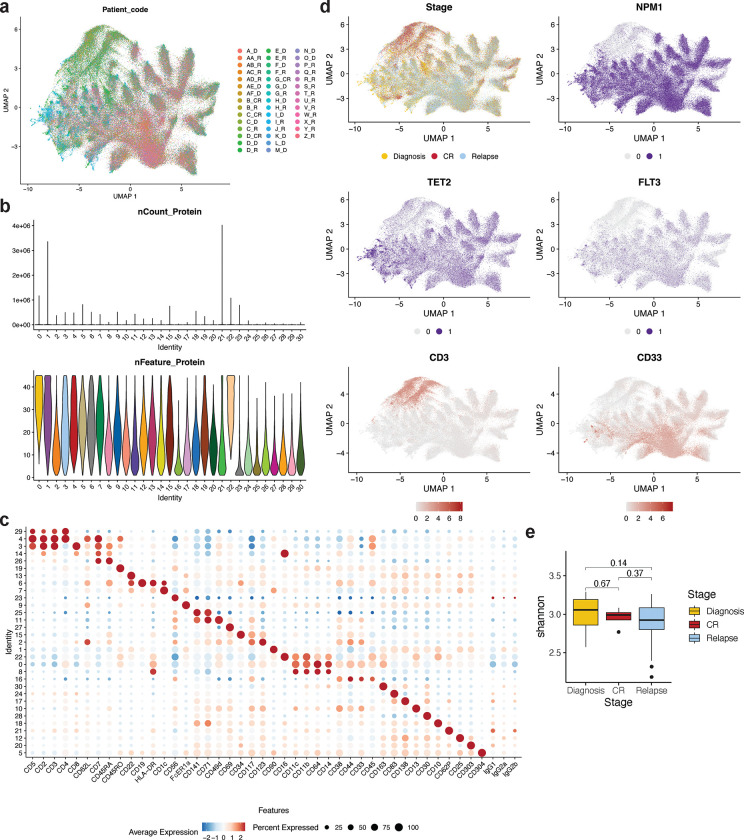
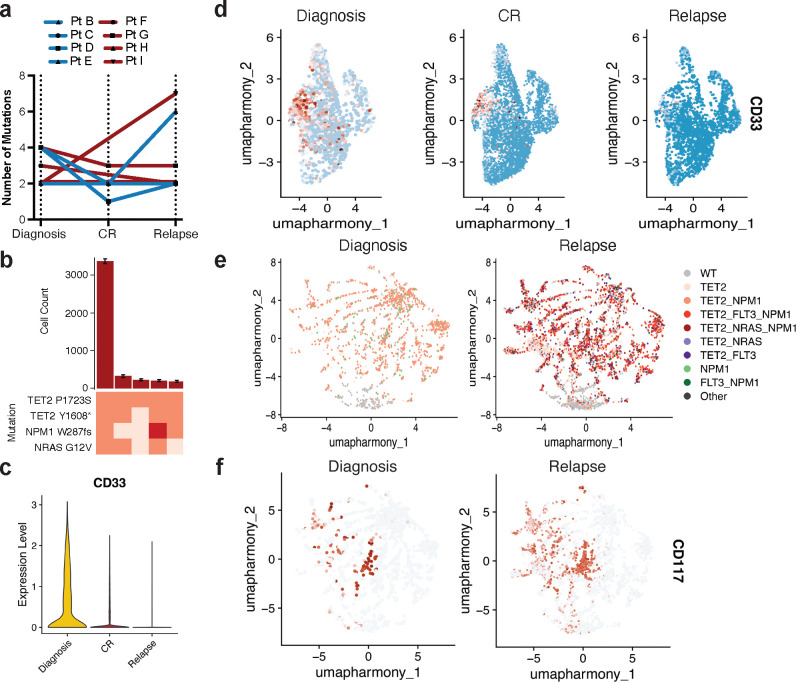
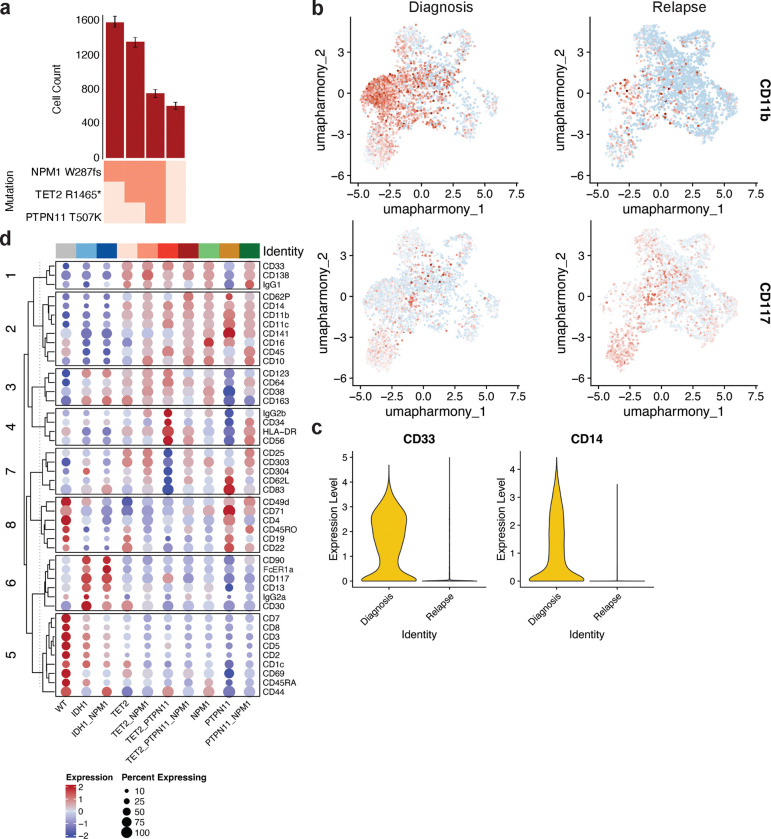
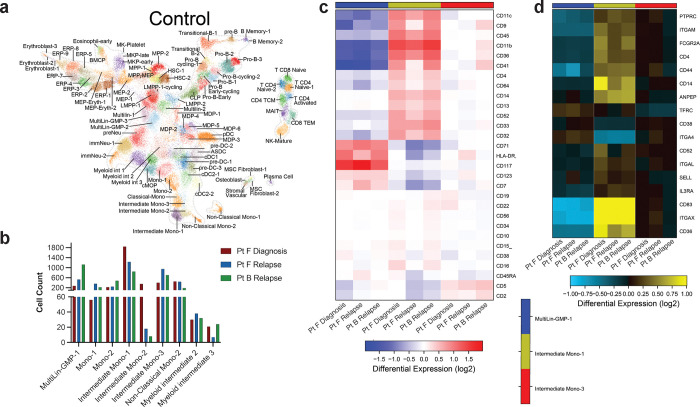
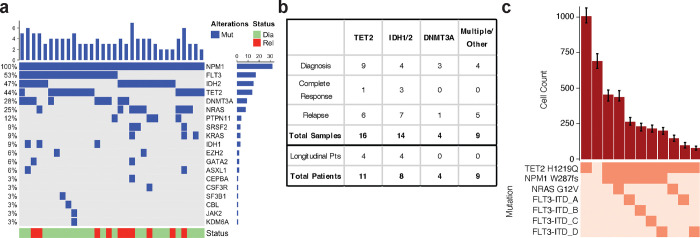
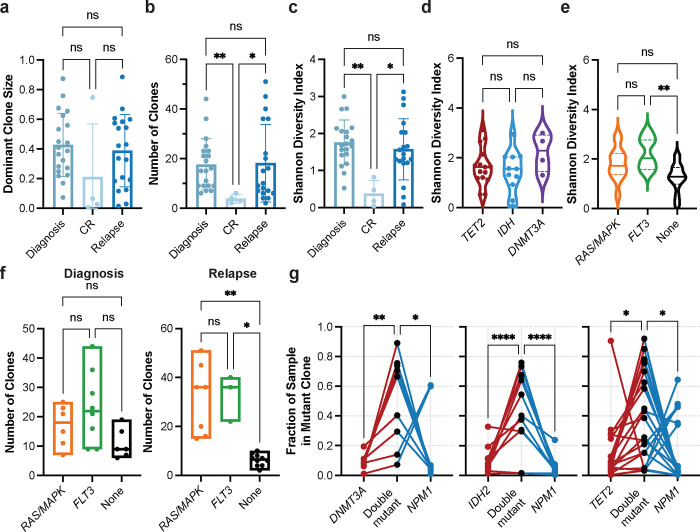
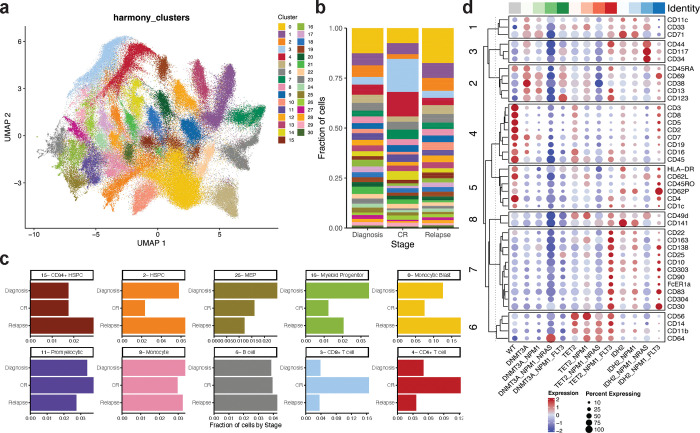
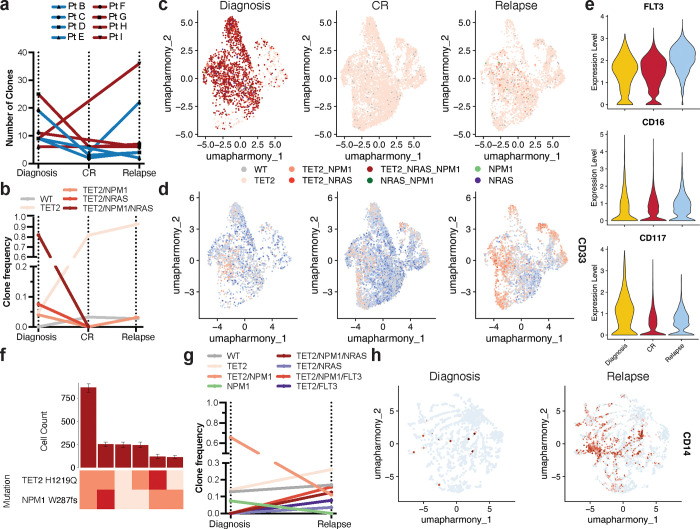
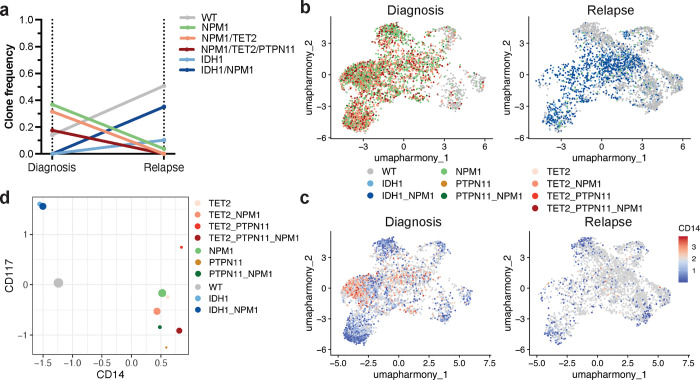
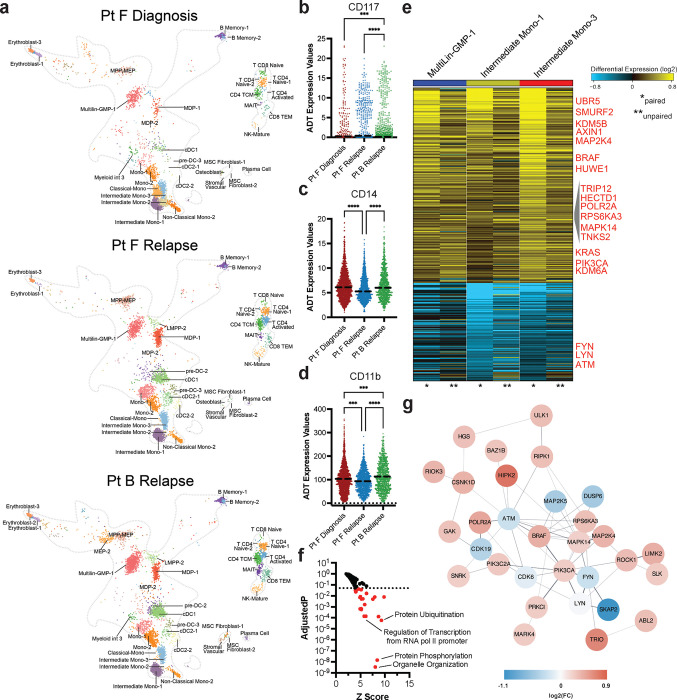
Acute myeloid leukemia (AML) is a multi-clonal disease, existing as a milieu of clones with unique but related genotypes as initiating clones acquire subsequent mutations. However, bulk sequencing cannot fully capture AML clonal architecture or the clonal evolution that occurs as patients undergo therapy. To interrogate clonal evolution, we performed simultaneous single cell molecular profiling and immunophenotyping on 43 samples from 32 NPM1-mutant AML patients at different stages of disease. Here we show that diagnosis and relapsed AML samples display similar clonal architecture patterns, but signaling mutations can drive increased clonal diversity specifically at relapse. We uncovered unique genotype-immunophenotype relationships regardless of disease state, suggesting leukemic lineage trajectories can be hard-wired by the mutations present. Analysis of longitudinal samples from patients on therapy identified dynamic clonal, transcriptomic, and immunophenotypic changes. Our studies provide resolved understanding of leukemic clonal evolution and the relationships between genotype and cell state in leukemia biology.
Conflict of interest statement
Competing Interests L.A.M. and R.L.B. had previously received honoraria for speaking arrangements and had previously served on a Speakers Bureau for Mission Bio, Inc. R.L.L. is on the supervisory board of QIAGEN and serves as a scientific advisor to Auron, Imago, Prelude, Zentalis Pharmaceuticals Mission Bio, Syndax, Ajax, Bakx, C4 Therapeutics and Isoplexis, for which he receives equity support. R.L.L. receives research support from Abbvie and Ajax, has served as a consultant for MorphoSys, Janssen, Incyte, and Novartis. J.C.B. has ownership interest in Vincera, an advisory and consultancy role with Novartis, Syndax, and Vincera, research funding from Genentech, Janssen, Acerta, and Pharmacyclics, an AbbVie company. R.L.L. has received honoraria from AstraZeneca and Incyte for invited lectures. A.-K.E. has received an honorarium from AstraZeneca for serving on their Diversity, Equity, and Inclusion Advisory Board and has received a research grant from Novartis. Spouse of A.-K.E. has ownership interest in Karyopharm Therapeutics. The other authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources