

This is a preprint.
Unveiling epigenetic regulatory elements associated with breast cancer development
- PMID: 39605637
- PMCID: PMC11601335
- DOI: 10.1101/2024.11.12.623187
Unveiling epigenetic regulatory elements associated with breast cancer development
Update in
-
Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development.Int J Mol Sci. 2025 Jul 8;26(14):6558. doi: 10.3390/ijms26146558. Int J Mol Sci. 2025. PMID: 40724805 Free PMC article.
Abstract
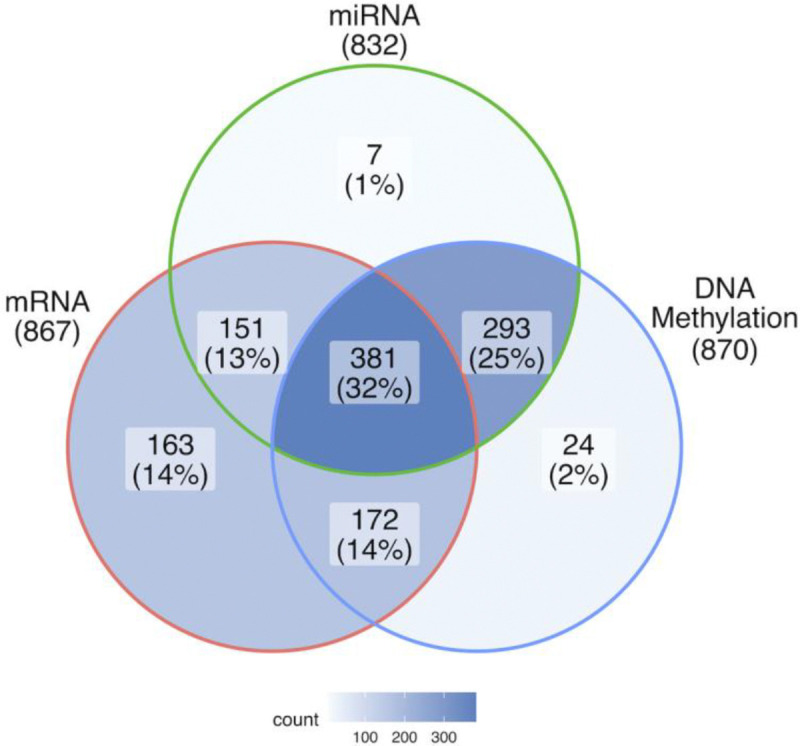
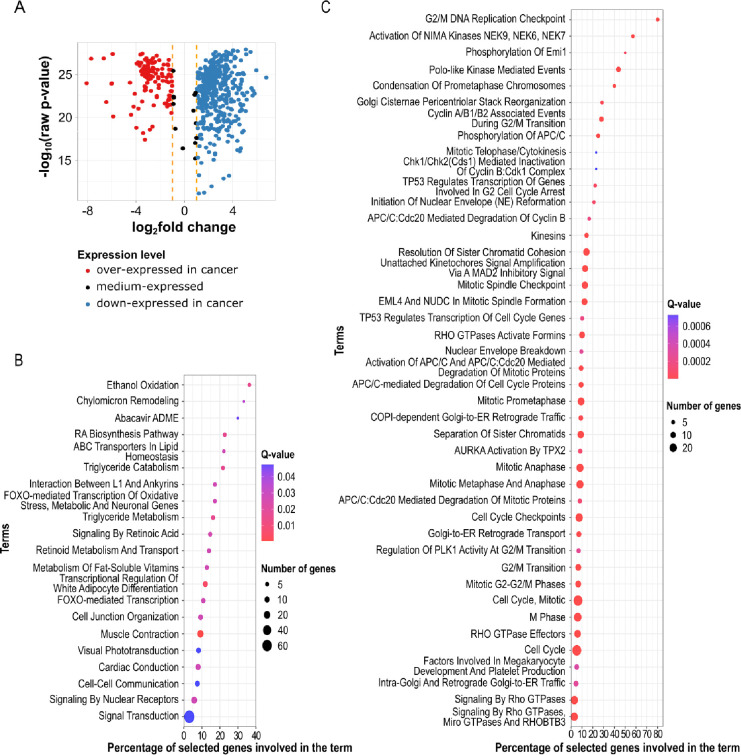
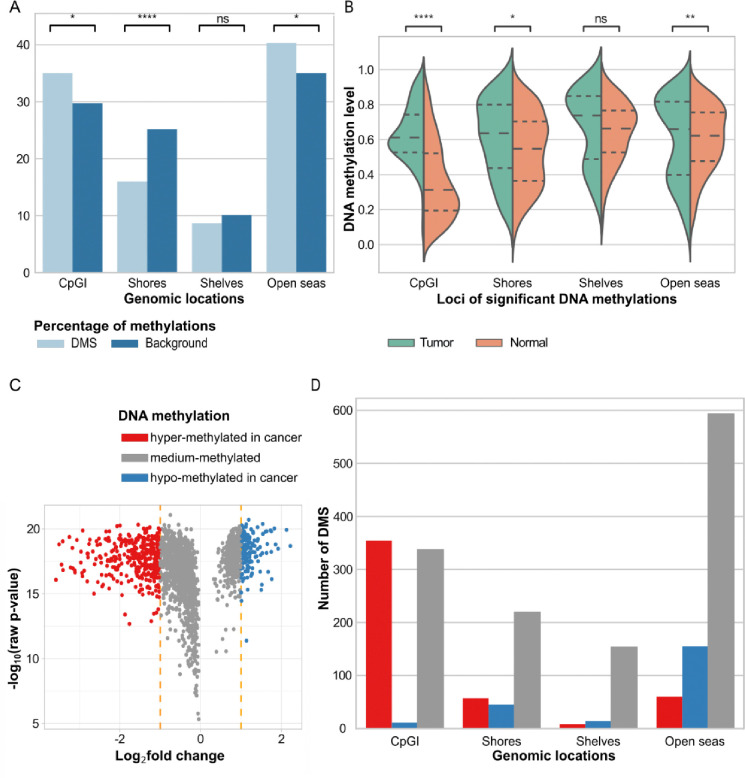
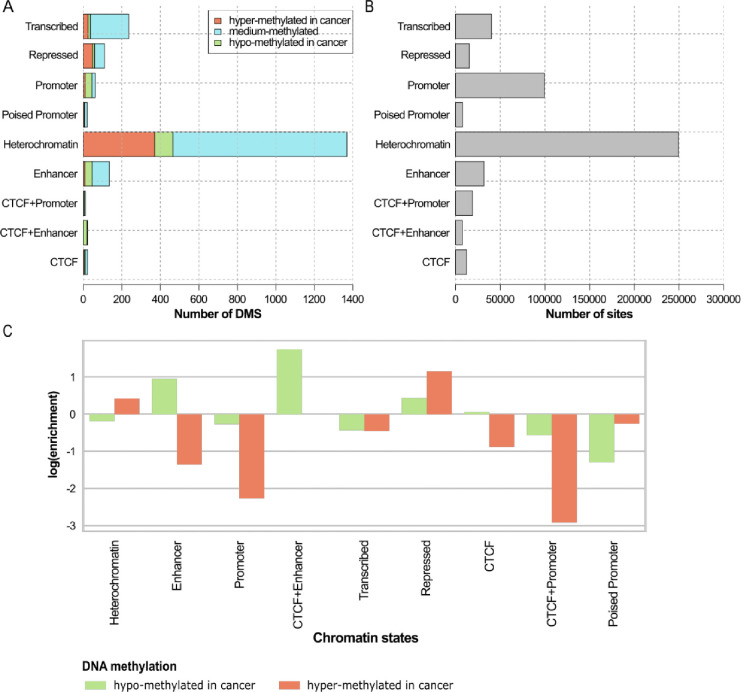
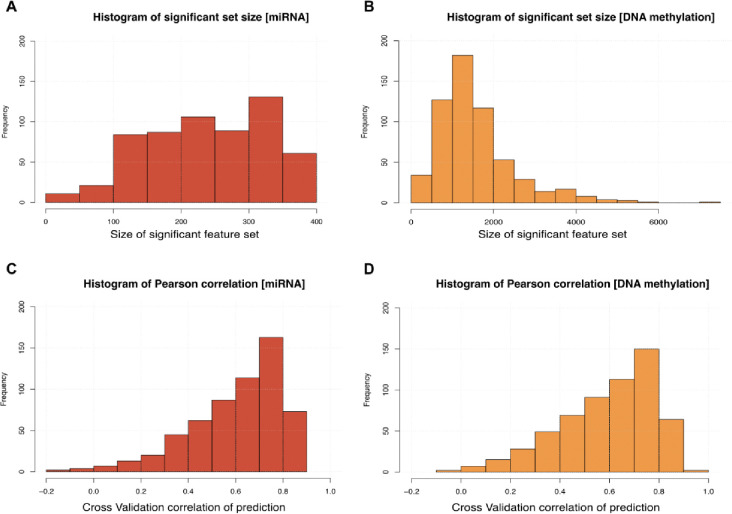
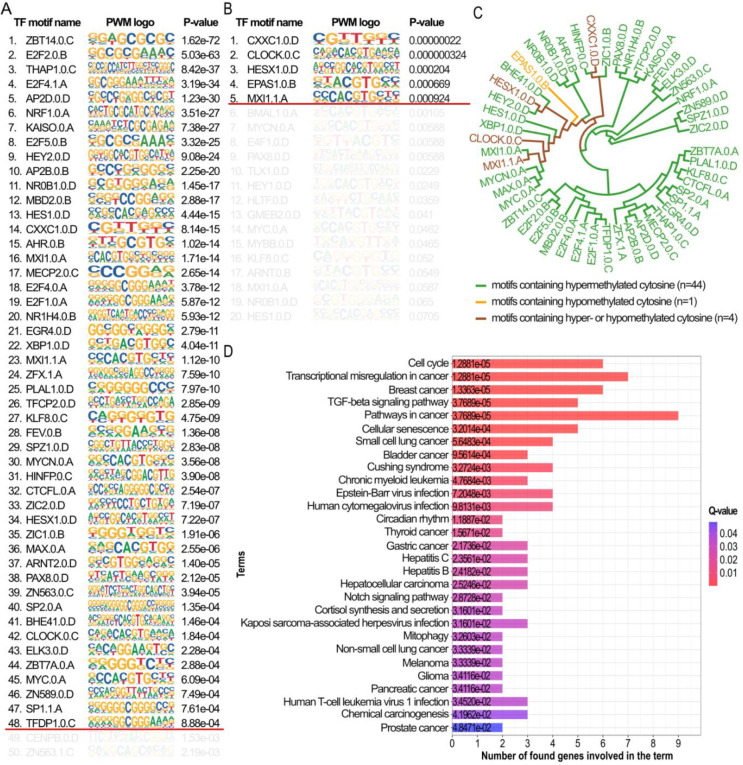
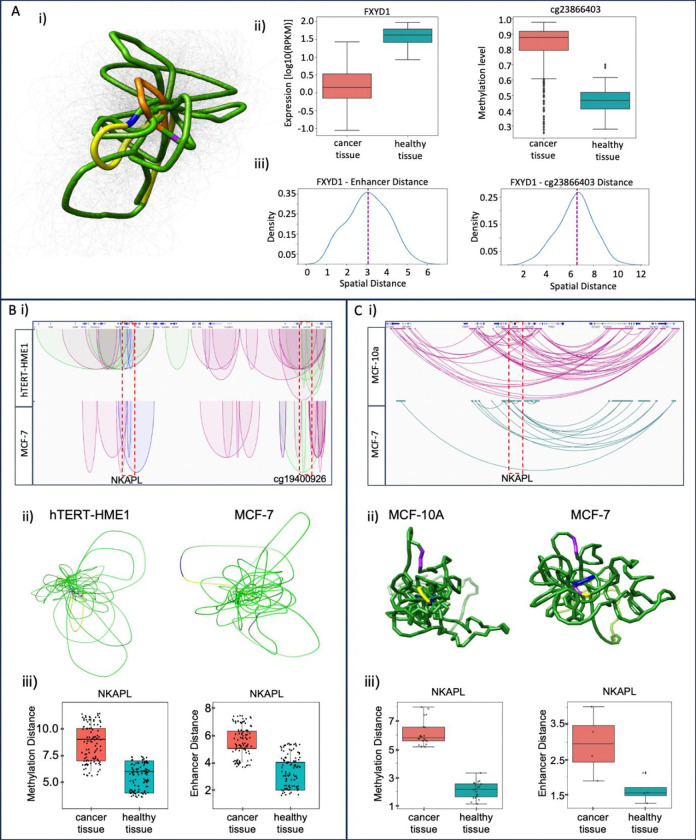
Breast cancer is the most common cancer in women and the 2nd most common cancer worldwide, yearly impacting over 2 million females and causing 650 thousand deaths. It has been widely studied, but its epigenetic variation is not entirely unveiled. We aimed to identify epigenetic mechanisms impacting the expression of breast cancer related genes to detect new potential biomarkers and therapeutic targets. We considered The Cancer Genome Atlas database with over 800 samples and several omics datasets such as mRNA, miRNA, DNA methylation, which we used to select 2701 features that were statistically significant to differ between cancer and control samples using the Monte Carlo Feature Selection and Interdependency Discovery algorithm, from an initial total of 417,486. Their biological impact on cancerogenesis was confirmed using: statistical analysis, natural language processing, linear and machine learning models as well as: transcription factors identification, drugs and 3D chromatin structure analyses. Classification of cancer vs control samples on the selected features returned high classification weighted Accuracy from 0.91 to 0.98 depending on feature-type: mRNA, miRNA, DNA methylation, and classification algorithm. In general, cancer samples showed lower expression of differentially expressed genes and increased β-values of differentially methylated sites. We identified mRNAs whose expression is well explained by miRNA expression and differentially methylated sites β-values. We recognized differentially methylated sites possibly affecting NRF1 and MXI1 transcription factors binding, causing a disturbance in NKAPL and PITX1 expression, respectively. Our 3D models showed more loosely packed chromatin in cancer. This study successfully points out numerous possible regulatory dependencies.
Keywords: MCFS-ID; MXI1; Monte Carlo Feature Selection; NKAPL; NLP; NRF1; PITX1; breast cancer; chromatin structure; differentially methylated sites; epigenetic regulation; transcription factor.
Figures



References
-
- Bean GR, Lin CY. Breast neuroendocrine neoplasms: practical applications and continuing challenges in the era of the 5th edition of the WHO classification of breast tumours. Diagnostic Histopathology. 2021. Jan;27(4):139–47. 10.1016/j.mpdhp.2021.01.001 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources