

This is a preprint.
Accurate de novo design of high-affinity protein binding macrocycles using deep learning
- PMID: 39605685
- PMCID: PMC11601608
- DOI: 10.1101/2024.11.18.622547
Accurate de novo design of high-affinity protein binding macrocycles using deep learning
Update in
-
Accurate de novo design of high-affinity protein-binding macrocycles using deep learning.Nat Chem Biol. 2025 Jun 20. doi: 10.1038/s41589-025-01929-w. Online ahead of print. Nat Chem Biol. 2025. PMID: 40542165
Abstract
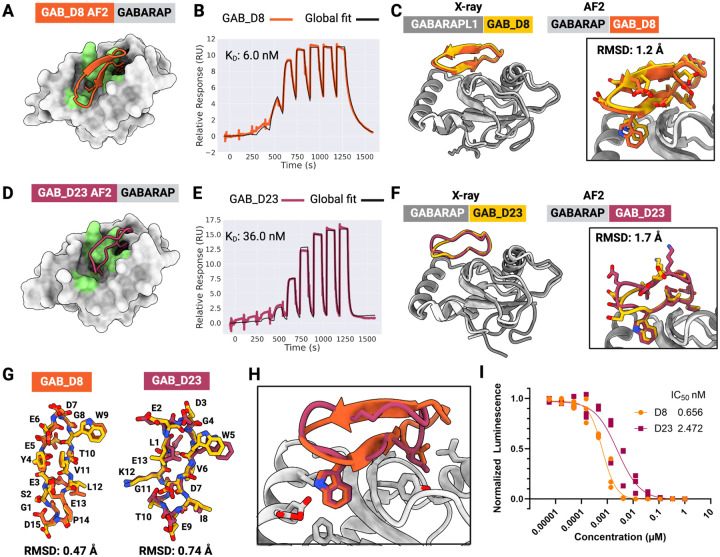
The development of macrocyclic binders to therapeutic proteins typically relies on large-scale screening methods that are resource-intensive and provide little control over binding mode. Despite considerable progress in physics-based methods for peptide design and deep-learning methods for protein design, there are currently no robust approaches for de novo design of protein-binding macrocycles. Here, we introduce RFpeptides, a denoising diffusion-based pipeline for designing macrocyclic peptide binders against protein targets of interest. We test 20 or fewer designed macrocycles against each of four diverse proteins and obtain medium to high-affinity binders against all selected targets. Designs against MCL1 and MDM2 demonstrate KD between 1-10 μM, and the best anti-GABARAP macrocycle binds with a KD of 6 nM and a sub-nanomolar IC50 in vitro. For one of the targets, RbtA, we obtain a high-affinity binder with KD < 10 nM despite starting from the target sequence alone due to the lack of an experimentally determined target structure. X-ray structures determined for macrocycle-bound MCL1, GABARAP, and RbtA complexes match very closely with the computational design models, with three out of the four structures demonstrating Ca RMSD of less than 1.5 Å to the design models. In contrast to library screening approaches for which determining binding mode can be a major bottleneck, the binding modes of RFpeptides-generated macrocycles are known by design, which should greatly facilitate downstream optimization. RFpeptides thus provides a powerful framework for rapid and custom design of macrocyclic peptides for diagnostic and therapeutic applications.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials