

This is a preprint.
Boltz-1 Democratizing Biomolecular Interaction Modeling
- PMID: 39605745
- PMCID: PMC11601547
- DOI: 10.1101/2024.11.19.624167
Boltz-1 Democratizing Biomolecular Interaction Modeling
Abstract
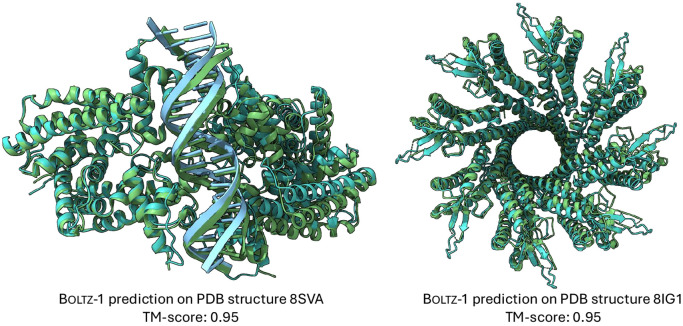
Understanding biomolecular interactions is fundamental to advancing fields like drug discovery and protein design. In this paper, we introduce Boltz-1, an open-source deep learning model incorporating innovations in model architecture, speed optimization, and data processing achieving Alphafold3-level accuracy in predicting the 3D structures of biomolecular complexes. Boltz-1 demonstrates a performance on-par with state-of-the-art commercial models on a range of diverse benchmarks, setting a new benchmark for commercially accessible tools in structural biology. Further, we push the boundary of capabilities of these models with Boltz-steering, a new inference time steering technique that is able to fix hallucinations and non-physical predictions from the models. By releasing the training and inference code, model weights, datasets, and benchmarks under the MIT open license, we aim to foster global collaboration, accelerate discoveries, and provide a robust platform for advancing biomolecular modeling.
Figures






References
-
- Ahdritz Gustaf, Bouatta Nazim, Floristean Christina, Kadyan Sachin, Xia Qinghui, Gerecke William, O’Donnell Timothy J, Berenberg Daniel, Fisk Ian, Zanichelli Niccolò, et al. Openfold: Retraining alphafold2 yields new insights into its learning mechanisms and capacity for generalization. Nature Methods, pages 1–11, 2024. - PMC - PubMed
-
- Bansal Arpit, Chu Hong-Min, Schwarzschild Avi, Sengupta Soumyadip, Goldblum Micah, Geiping Jonas, and Goldstein Tom. Universal guidance for diffusion models. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, pages 843–852, 2023.
-
- Biasini Marco, Schmidt Tobias, Bienert Stefan, Mariani Valerio, Studer Gabriel, Haas Jürgen, Johner Niklaus, Schenk Andreas Daniel, Philippsen Ansgar, and Schwede Torsten. Openstructure: an integrated software framework for computational structural biology. Acta Crystallographica Section D: Biological Crystallography, 69(5):701–709, 2013. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources