

UHRF1 ubiquitin ligase activity supports the maintenance of low-density CpG methylation
- PMID: 39607687
- PMCID: PMC11662662
- DOI: 10.1093/nar/gkae1105
UHRF1 ubiquitin ligase activity supports the maintenance of low-density CpG methylation
Abstract
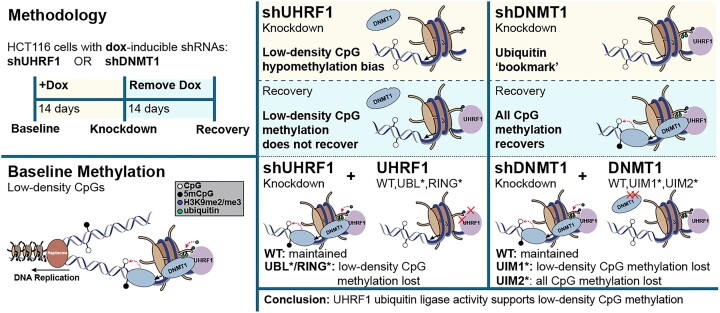
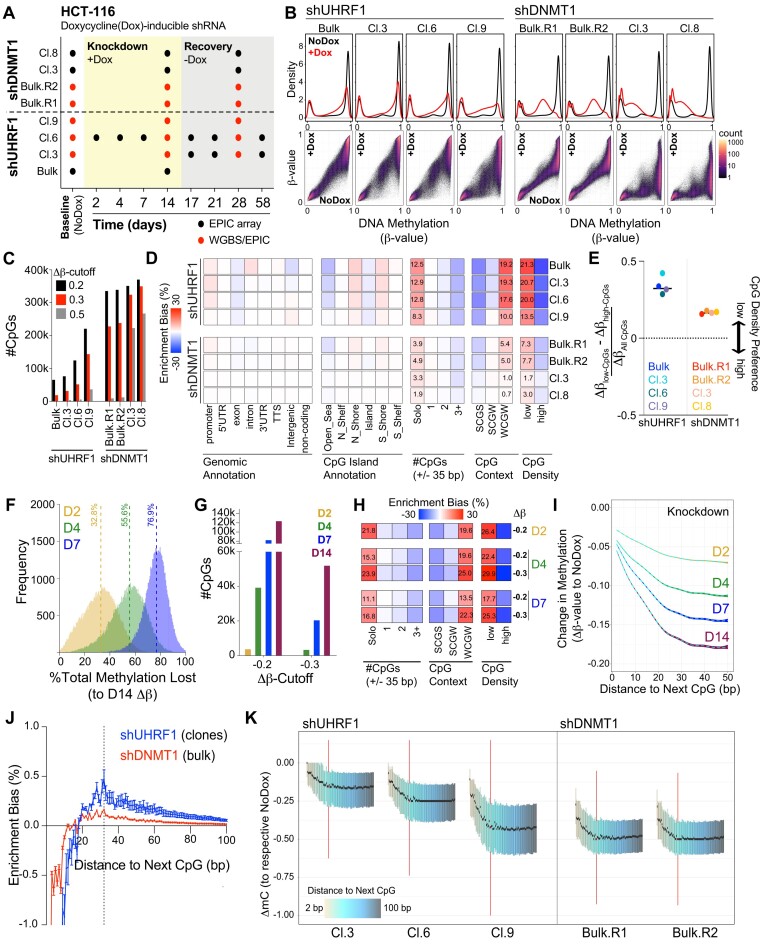
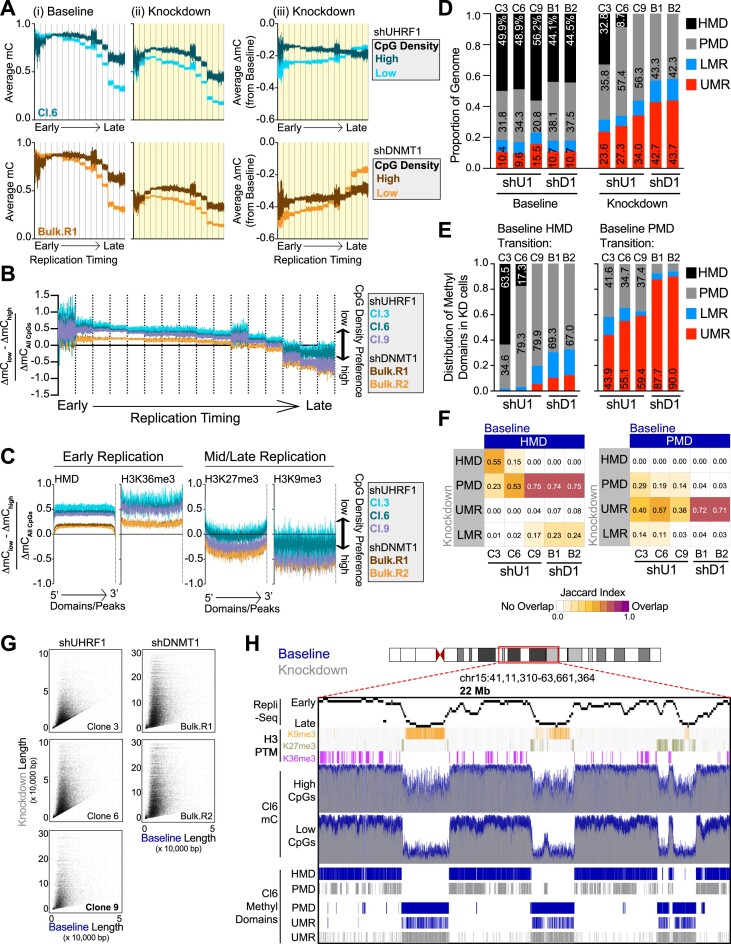
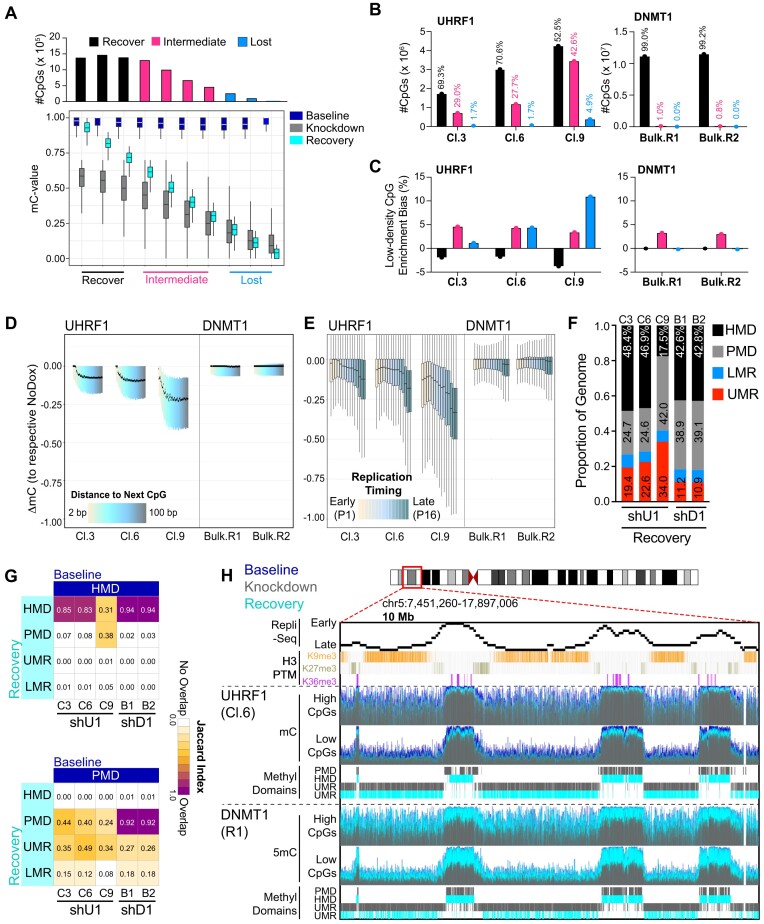
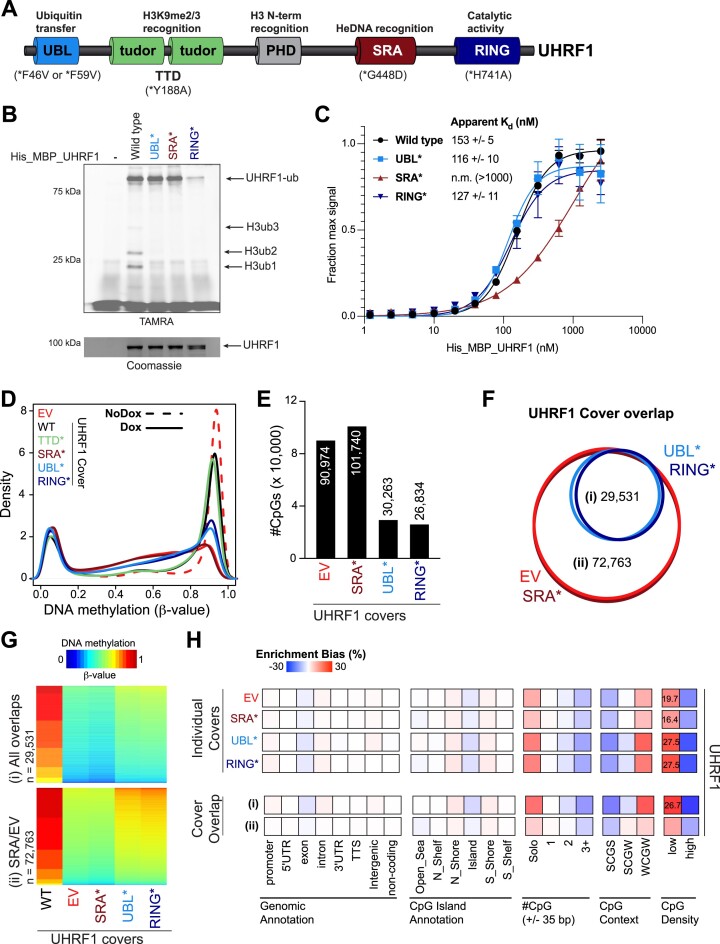
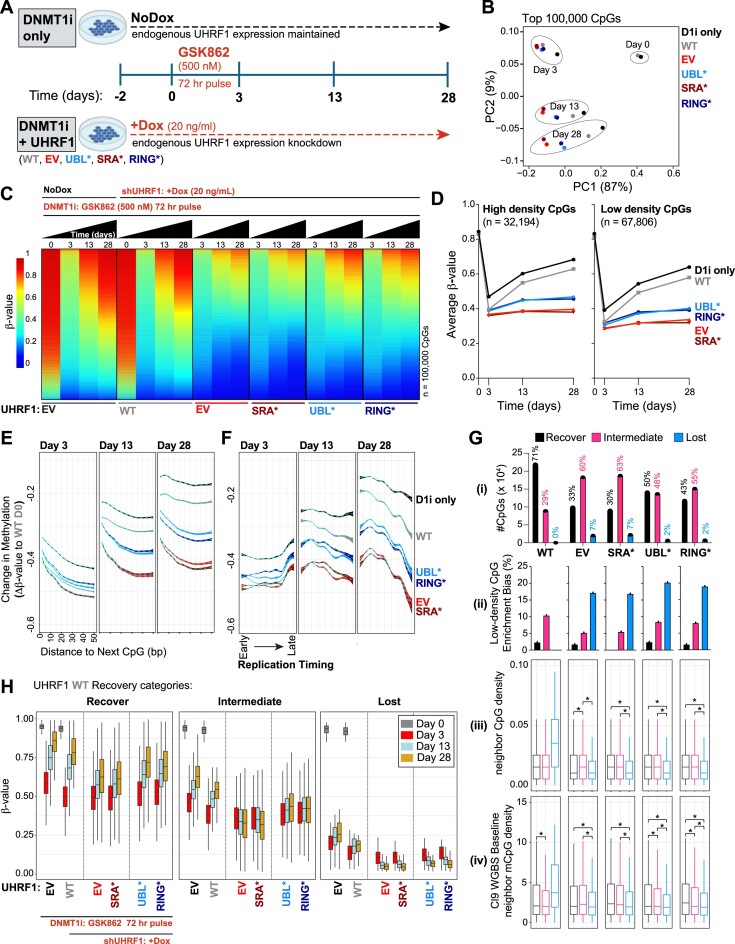
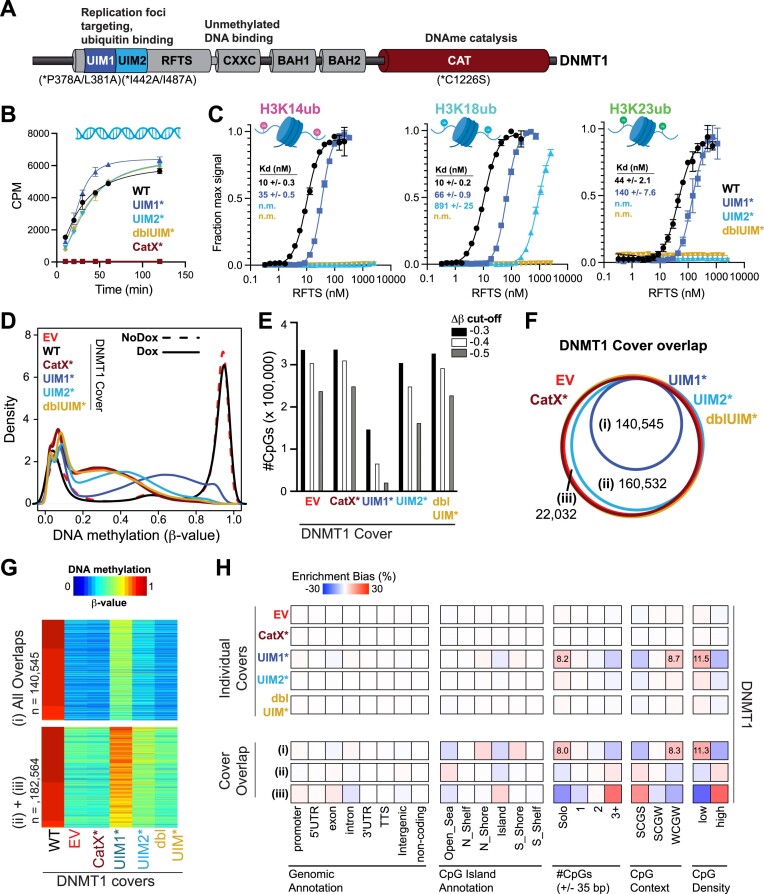
The RING E3 ubiquitin ligase UHRF1 is an established cofactor for DNA methylation inheritance. The model posits that nucleosomal engagement through histone and DNA interactions directs UHRF1 ubiquitin ligase activity toward lysines on histone H3 tails, creating binding sites for DNMT1 through ubiquitin interacting motifs (UIM1 and UIM2). However, the extent to which DNMT1 relies on ubiquitin signaling through UHRF1 in support of DNA methylation maintenance remains unclear. Here, with integrative epigenomic and biochemical analyses, we reveal that DNA methylation maintenance at low-density cytosine-guanine dinucleotides (CpGs) is particularly vulnerable to disruption of UHRF1 ubiquitin ligase activity and DNMT1 ubiquitin reading activity through UIM1. Hypomethylation of low-density CpGs in this manner induces formation of partially methylated domains (PMDs), a methylation signature observed across human cancers. In contrast, UIM2 disruption completely abolishes the DNA methylation maintenance function of DNMT1 in a CpG density-independent manner. In the context of DNA methylation recovery following acute DNMT1 depletion, we further reveal a 'bookmarking' function for UHRF1 ubiquitin ligase activity in support of DNA re-methylation. Collectively, these studies show that DNMT1-dependent DNA methylation inheritance is a ubiquitin-regulated process that is partially reliant on UHRF1 and suggest a disrupted UHRF1-DNMT1 ubiquitin signaling axis contributes to PMD formation in cancers.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Update of
-
UHRF1 ubiquitin ligase activity supports the maintenance of low-density CpG methylation.bioRxiv [Preprint]. 2024 Feb 16:2024.02.13.580169. doi: 10.1101/2024.02.13.580169. bioRxiv. 2024. Update in: Nucleic Acids Res. 2024 Dec 11;52(22):13733-13756. doi: 10.1093/nar/gkae1105. PMID: 38405904 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
