

Machine learning-enhanced immunopeptidomics applied to T-cell epitope discovery for COVID-19 vaccines
- PMID: 39609459
- PMCID: PMC11604954
- DOI: 10.1038/s41467-024-54734-9
Machine learning-enhanced immunopeptidomics applied to T-cell epitope discovery for COVID-19 vaccines
Abstract
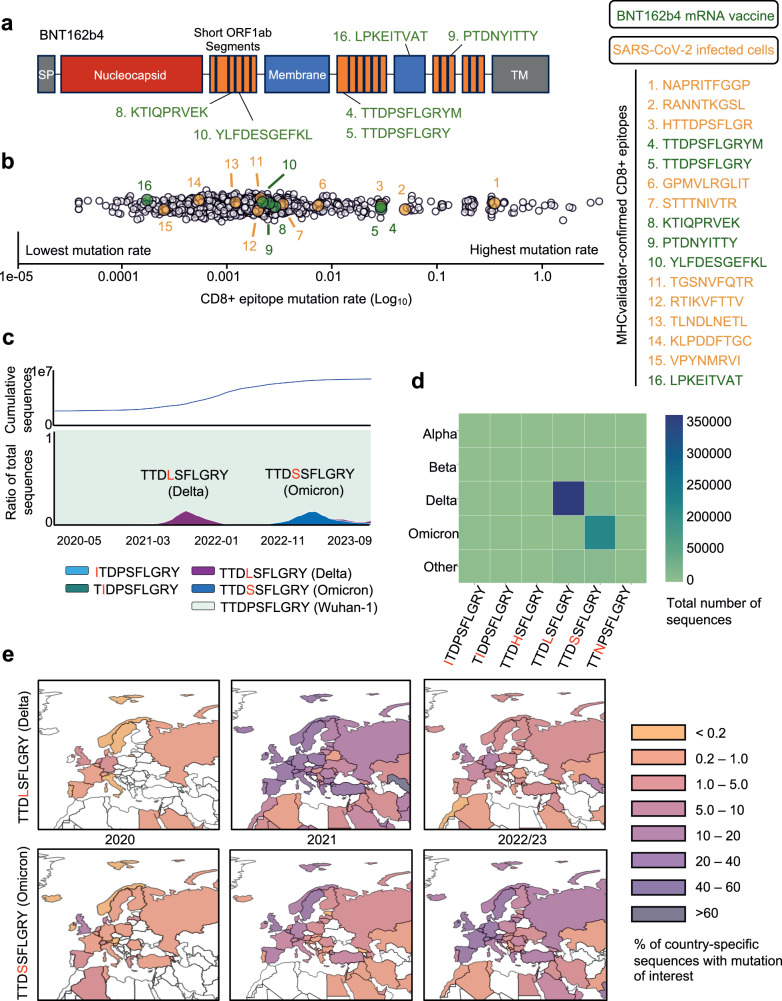
Next-generation T-cell-directed vaccines for COVID-19 focus on establishing lasting T-cell immunity against current and emerging SARS-CoV-2 variants. Precise identification of conserved T-cell epitopes is critical for designing effective vaccines. Here we introduce a comprehensive computational framework incorporating a machine learning algorithm-MHCvalidator-to enhance mass spectrometry-based immunopeptidomics sensitivity. MHCvalidator identifies unique T-cell epitopes presented by the B7 supertype, including an epitope from a + 1-frameshift in a truncated Spike antigen, supported by ribosome profiling. Analysis of 100,512 COVID-19 patient proteomes shows Spike antigen truncation in 0.85% of cases, revealing frameshifted viral antigens at the population level. Our EpiTrack pipeline tracks global mutations of MHCvalidator-identified CD8 + T-cell epitopes from the BNT162b4 vaccine. While most vaccine epitopes remain globally conserved, an immunodominant A*01-associated epitope mutates in Delta and Omicron variants. This work highlights SARS-CoV-2 antigenic features and emphasizes the importance of continuous adaptation in T-cell vaccine development.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: E.C. and I.S. are co-founders of Neomabs Biotechnologies Inc. PT is a co-founder of Epitopea. A.S. is a consultant for Alcimed, Gritstone, Darwin Health, EmerVax, Gilead Sciences, Guggenheim Securities, Link University, RiverVest Venture Partners, and Arcturus. La Jolla Institute for Immunology has filed for patent protection for various aspects of T cell epitope and vaccine design work. All other authors declare no competing interests. Inclusion & Ethics Statement: This research prioritizes inclusivity by engaging diverse populations, particularly underrepresented groups. All participants provided informed consent, and their confidentiality was safeguarded in accordance with ethical guidelines. The contributions of all team members were acknowledged, and potential biases were actively addressed. We aim to conduct research that advances knowledge while respecting the rights and dignity of all individuals involved.
Figures







References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
