

Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies
- PMID: 39609616
- PMCID: PMC11604929
- DOI: 10.1038/s42003-024-07290-3
Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies
Abstract
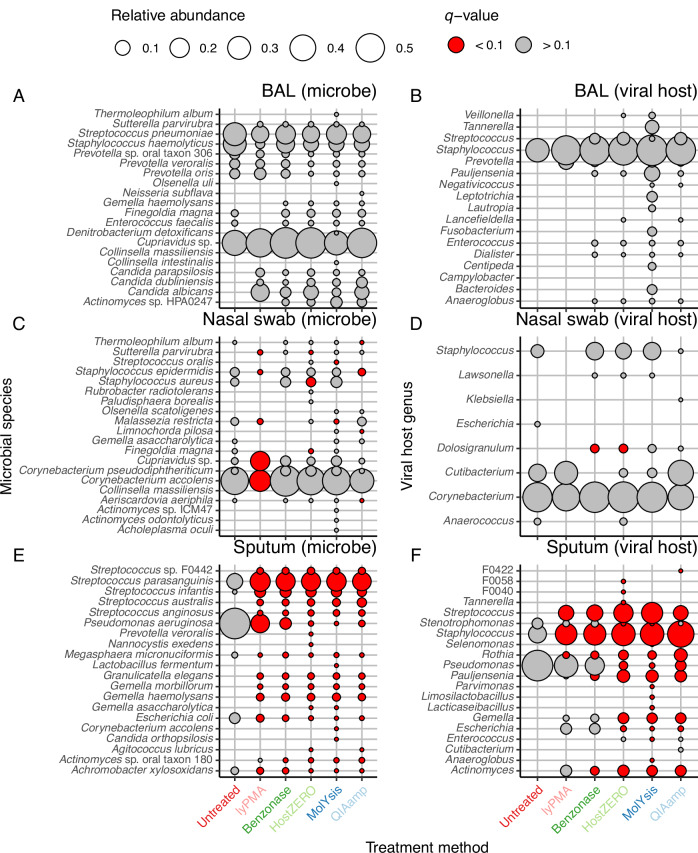
Most respiratory microbiome studies use amplicon sequencing due to high host DNA. Metagenomics sequencing offers finer taxonomic resolution, phage assessment, and functional characterization. We evaluated five host DNA depletion methods on frozen nasal swabs from healthy adults, sputum from people with cystic fibrosis (pwCF), and bronchoalveolar lavage (BAL) from critically ill patients. Median sequencing depth was 76.4 million reads per sample. Untreated nasal, sputum, and BAL had 94.1%, 99.2%, and 99.7% host reads, respectively. Host depletion effects varied by sample type, generally increasing microbial reads, species and functional richness; this was mediated by higher effective sequencing depth. Rarefaction curves showed species richness saturation at 0.5-2 million microbial reads. Most methods did not change Morisita-Horn dissimilarity for BAL and nasal samples although the proportion of gram-negative bacteria decreased for sputum from pwCF. Freezing did not affect the viability of Staphylococcus aureus but reduced the viability of Pseudomonas aeruginosa and Enterobacter spp.; this was mitigated by adding a cryoprotectant. QIAamp-based host depletion minimally impacted gram-negative viability even in non-cryoprotected frozen isolates. While some host depletion methods may shift microbial composition, metagenomics sequencing without host depletion severely underestimates microbial diversity of respiratory samples due to shallow effective sequencing depth and is not recommended.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: All ethical regulations relevant to human research participants were followed. Ethical approval for this study was obtained by the Institutional Review Board of Mass General Brigham (Protocol #2018P002934, 2019P002868 and 2020P001761).
Figures
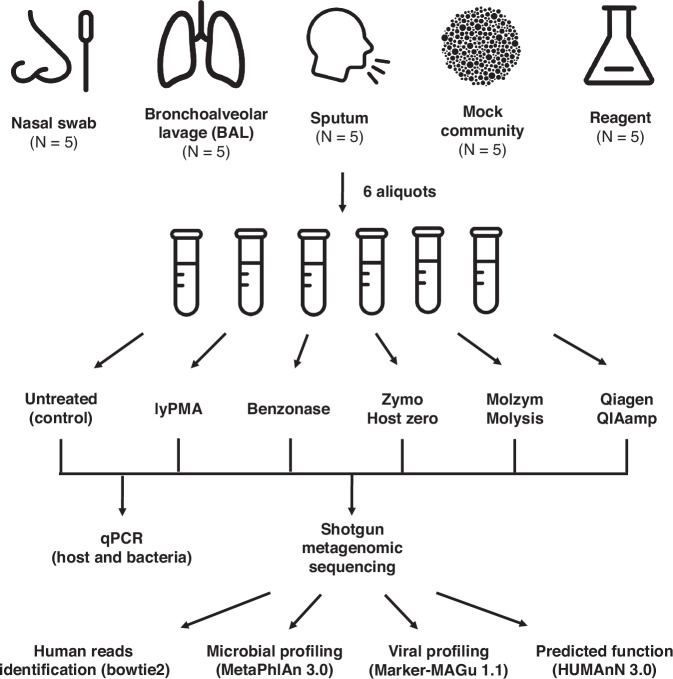
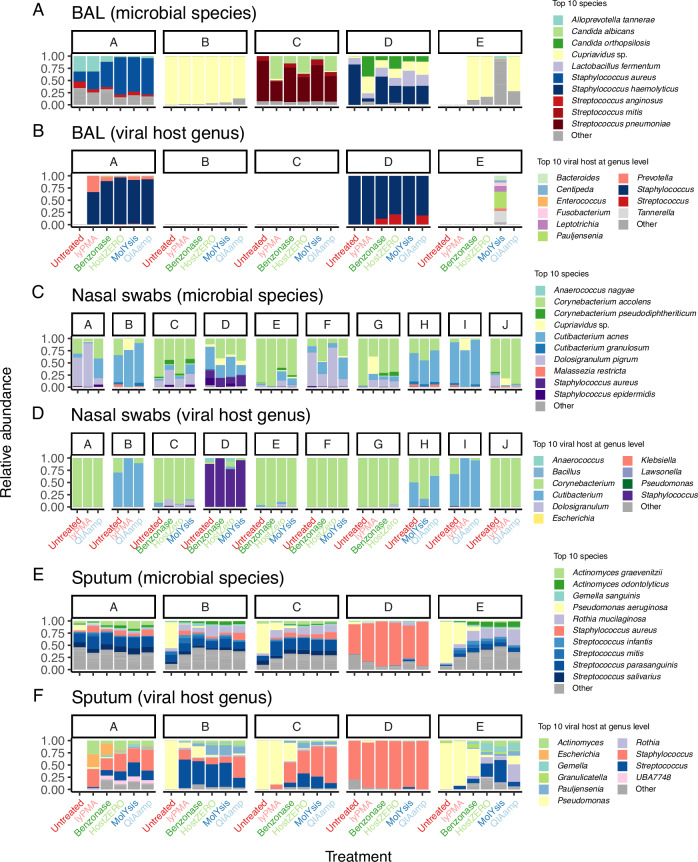
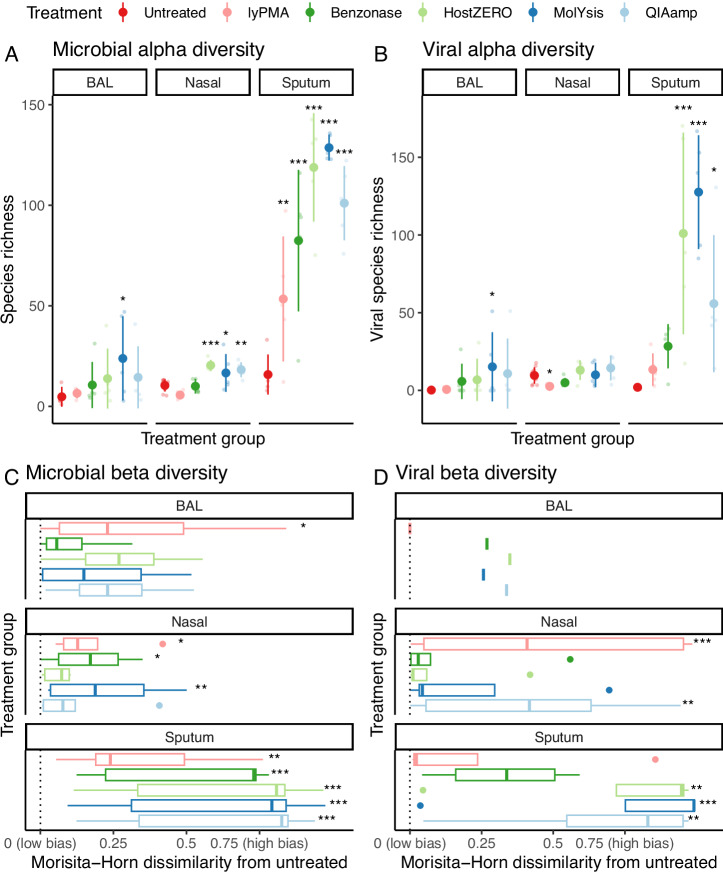
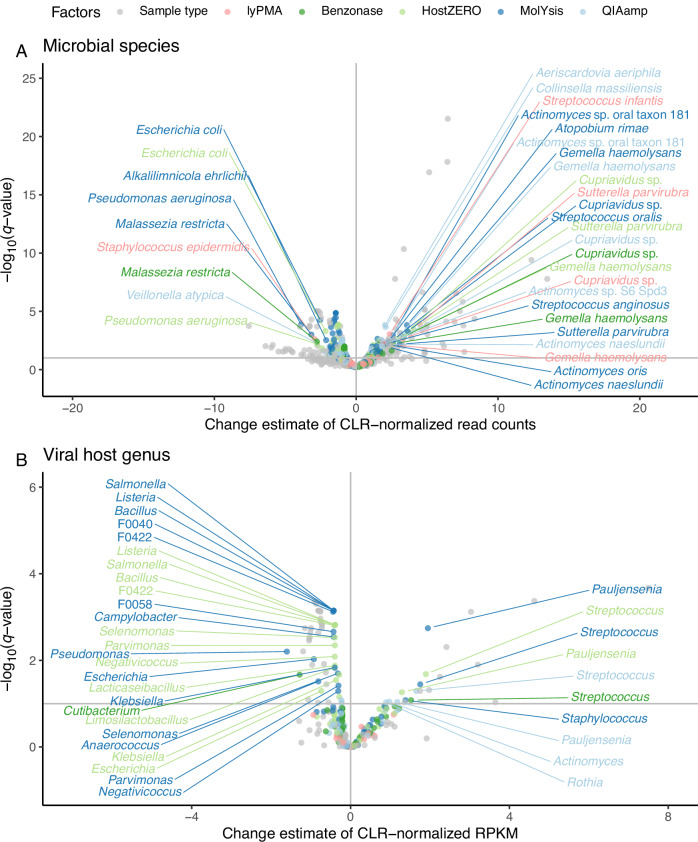

Update of
-
Host DNA depletion on frozen human respiratory samples enables successful metagenomic sequencing for microbiome studies.Res Sq [Preprint]. 2024 Jan 23:rs.3.rs-3638876. doi: 10.21203/rs.3.rs-3638876/v1. Res Sq. 2024. Update in: Commun Biol. 2024 Nov 28;7(1):1590. doi: 10.1038/s42003-024-07290-3. PMID: 38343829 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
