

Evolutionary genomics of Leishmania braziliensis across the neotropical realm
- PMID: 39609617
- PMCID: PMC11605123
- DOI: 10.1038/s42003-024-07278-z
Evolutionary genomics of Leishmania braziliensis across the neotropical realm
Abstract
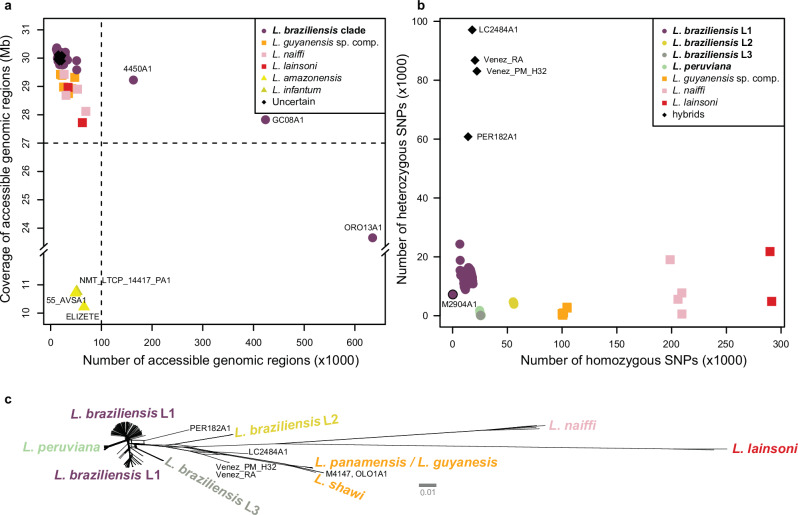
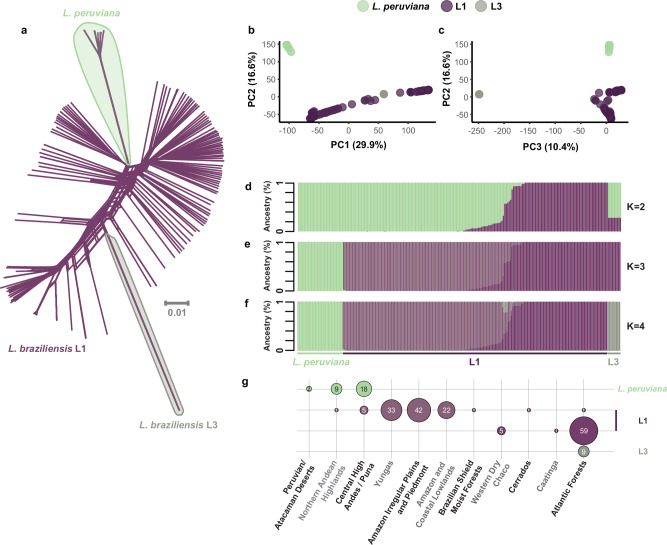
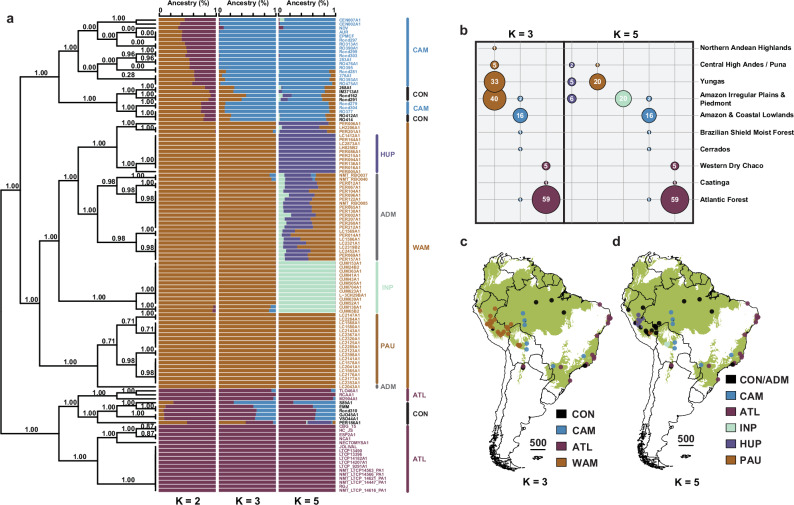
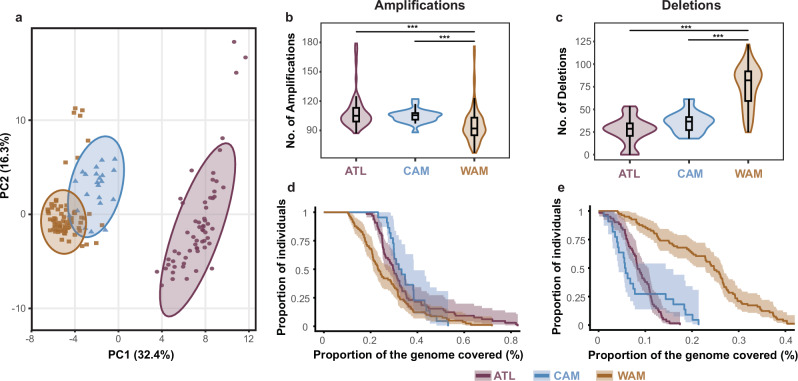
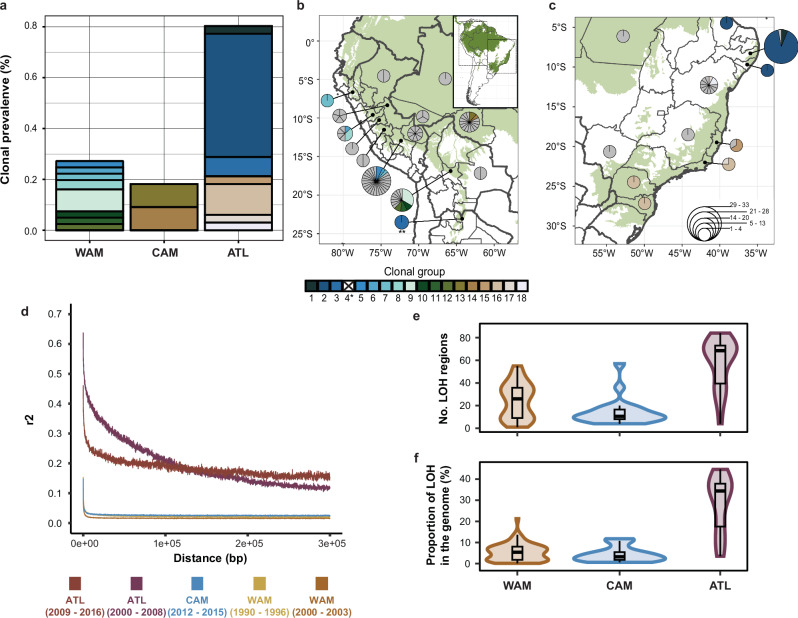
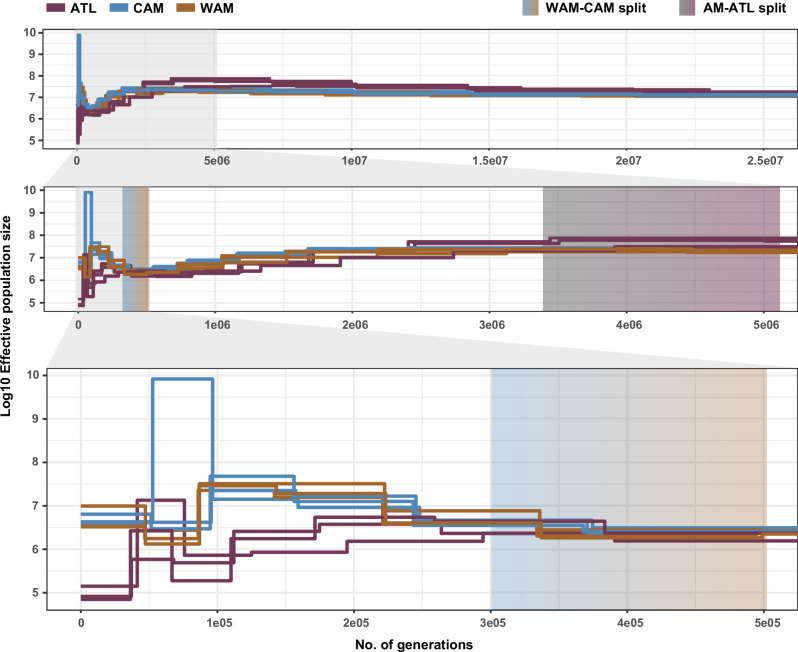
The Neotropical realm, one of the most biodiverse regions on Earth, houses a broad range of zoonoses that pose serious public health threats. Protozoan parasites of the Leishmania (Viannia) braziliensis clade cause zoonotic leishmaniasis in Latin America with clinical symptoms ranging from simple cutaneous to destructive, disfiguring mucosal lesions. We present the first comprehensive genome-wide continental study including 257 cultivated isolates representing most of the geographical distribution of this major human pathogen. The L. braziliensis clade is genetically highly heterogeneous, consisting of divergent parasite groups that are associated with different environments and vary greatly in diversity. Apart from several small ecologically isolated groups with little diversity, our sampling identifies two major parasite groups, one associated with the Amazon and the other with the Atlantic Forest biomes. These groups show different recombination histories, as suggested by high levels of heterozygosity and effective population sizes in the Amazonian group in contrast to high levels of linkage and clonality in the Atlantic group. We argue that these differences are linked to strong eco-epidemiological differences between the two regions. In contrast to geographically focused studies, our study provides a broad understanding of the molecular epidemiology of zoonotic parasites circulating in tropical America.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

References
MeSH terms
Grants and funding
- G092921N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- 1226120N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- 206194/WT_/Wellcome Trust/United Kingdom
- Finance Code 001/Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brazilian Federal Agency for the Support and Evaluation of Graduate Education)
- 302622/2017-9/Ministry of Science, Technology and Innovation | Conselho Nacional de Desenvolvimento Científico e Tecnológico (National Council for Scientific and Technological Development)
- E26-202.569/2019/Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro)
- E_ 08/2020 -COLBIO-210.285/2021/Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro)
- E-26/205.730/2022/Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro)
- 205.731/2022/Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro)
LinkOut - more resources
Full Text Sources
