

Understanding the role of TNFR2 signaling in the tumor microenvironment of breast cancer
- PMID: 39609700
- PMCID: PMC11603874
- DOI: 10.1186/s13046-024-03218-1
Understanding the role of TNFR2 signaling in the tumor microenvironment of breast cancer
Abstract
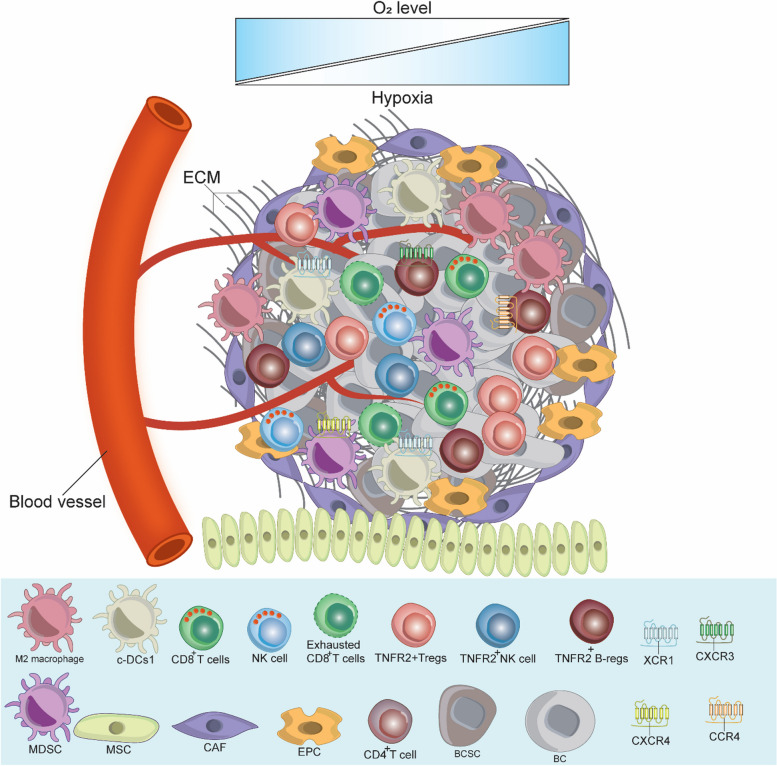
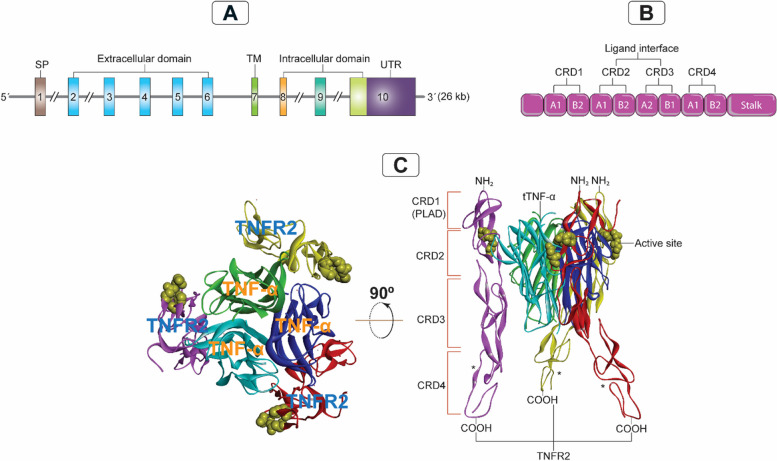
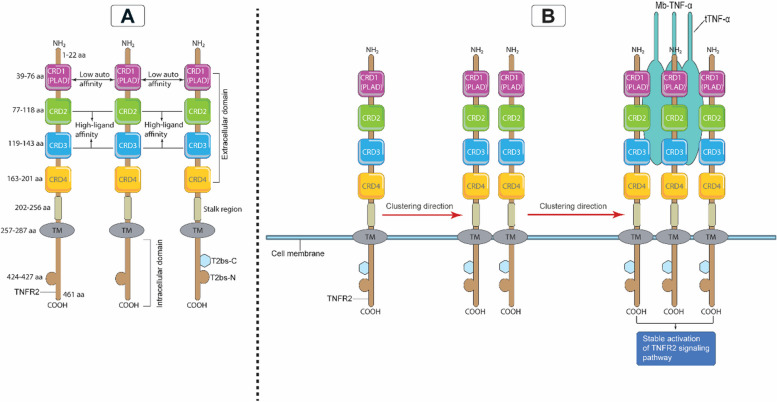
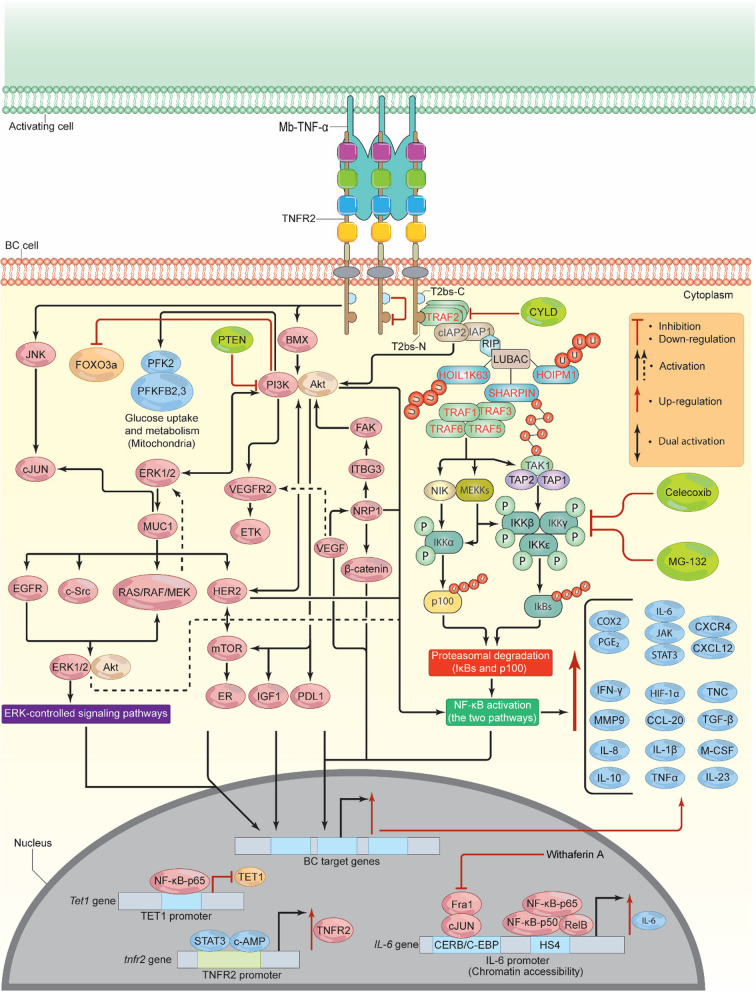
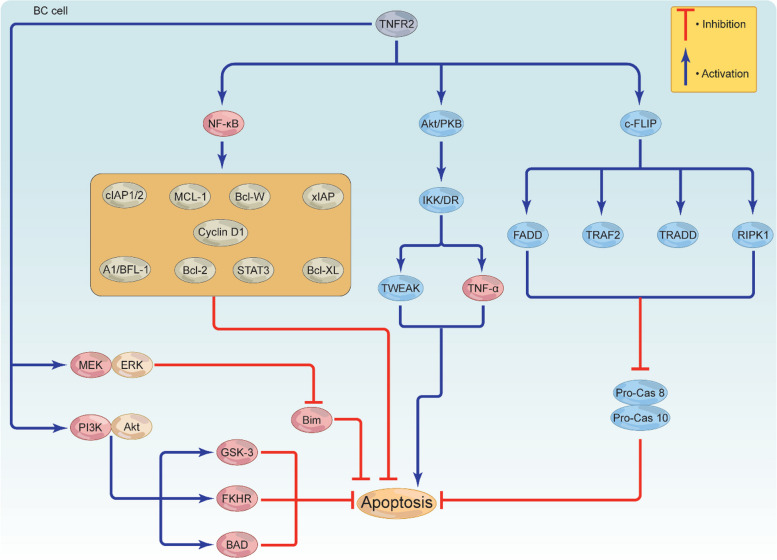
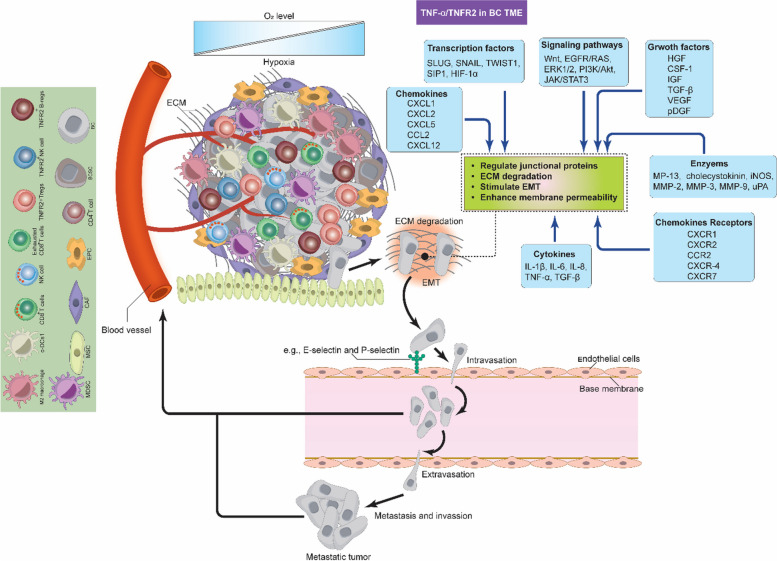
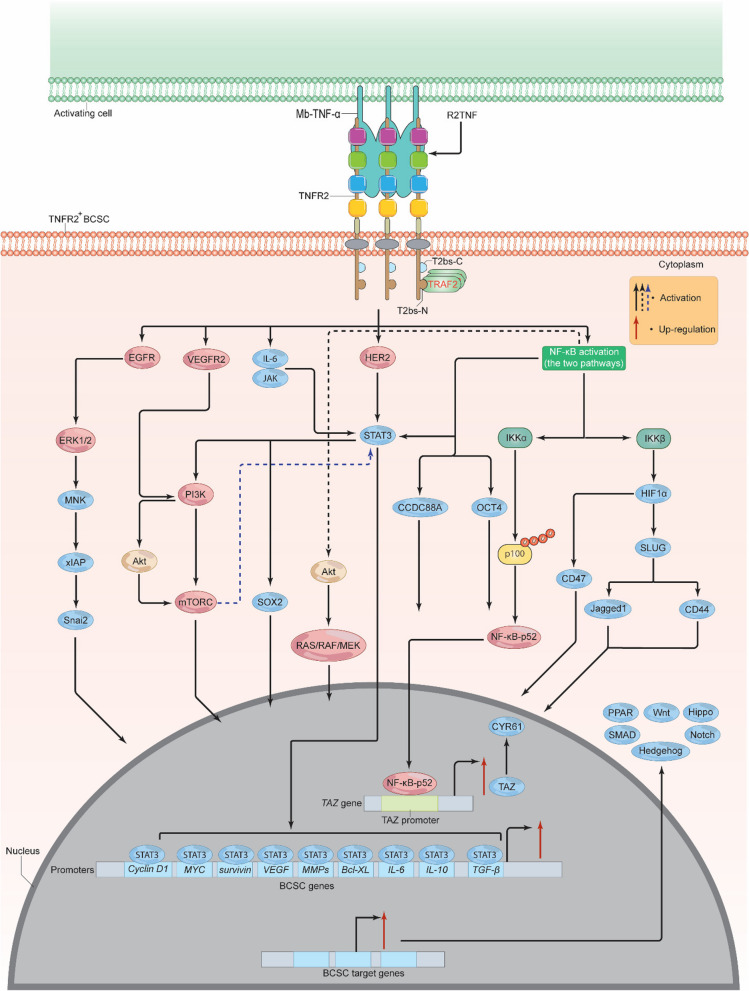
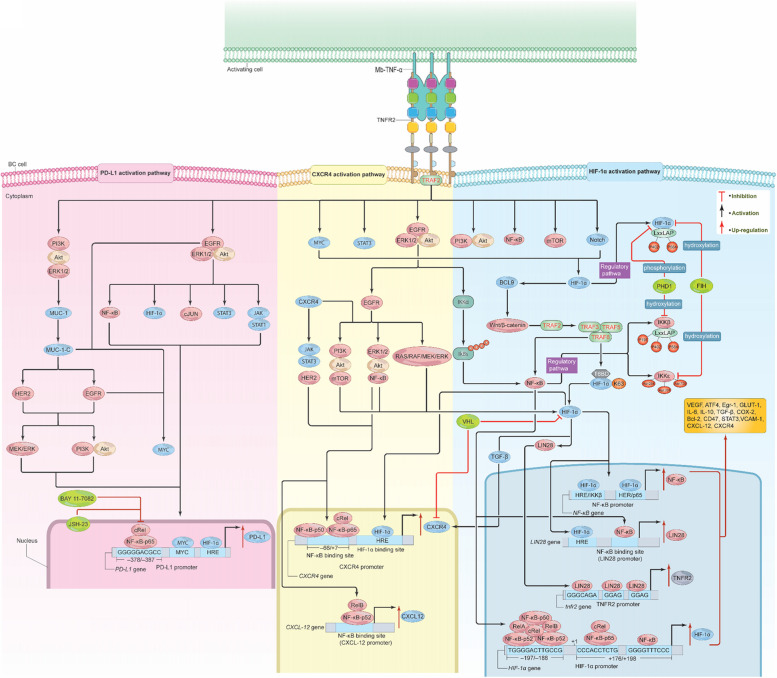
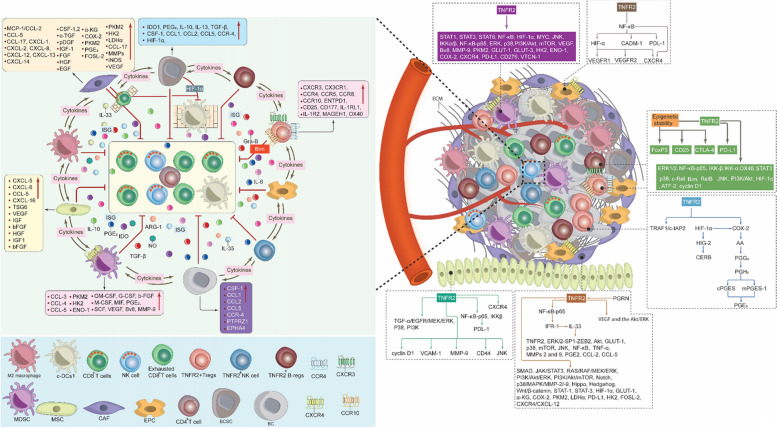
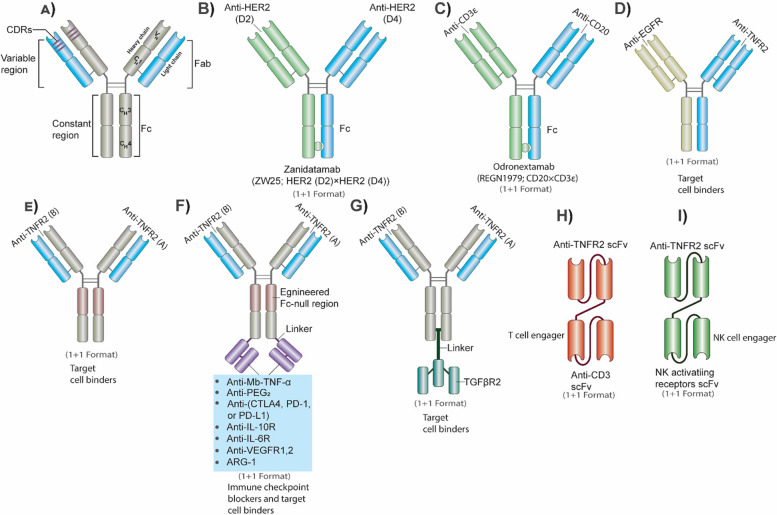
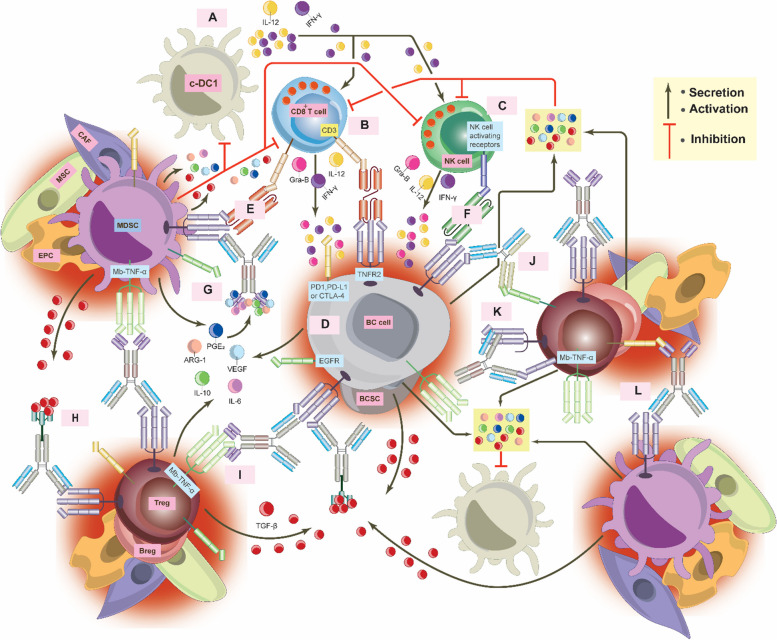
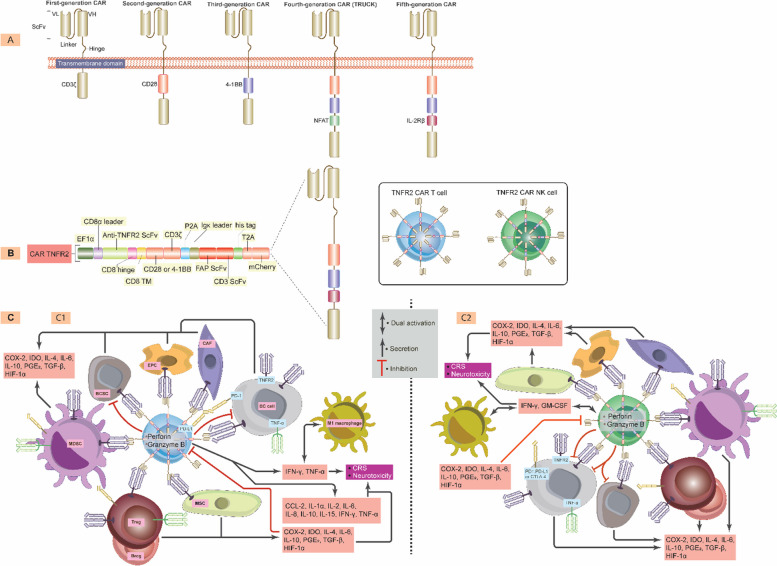
Breast cancer (BC) is the most frequently diagnosed malignancy among women. It is characterized by a high level of heterogeneity that emerges from the interaction of several cellular and soluble components in the tumor microenvironment (TME), such as cytokines, tumor cells and tumor-associated immune cells. Tumor necrosis factor (TNF) receptor 2 (TNFR2) appears to play a significant role in microenvironmental regulation, tumor progression, immune evasion, drug resistance, and metastasis of many types of cancer, including BC. However, the significance of TNFR2 in BC biology is not fully understood. This review provides an overview of TNFR2 biology, detailing its activation and its interactions with important signaling pathways in the TME (e.g., NF-κB, MAPK, and PI3K/Akt pathways). We discuss potential therapeutic strategies targeting TNFR2, with the aim of enhancing the antitumor immune response to BC. This review provides insights into role of TNFR2 as a major immune checkpoint for the future treatment of patients with BC.
Keywords: CD120b; Immune checkpoint; Immunosuppressive TME; TNF; TNFRSF1B.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declared that they have no competing interests.
Figures
References
-
- Sung, H., J. Ferlay, R.L. Siegel, et al.1, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians, 2021. 71(3): p. 209–249. 10.3322/caac.21660. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
