

Paired analysis of host and pathogen genomes identifies determinants of human tuberculosis
- PMID: 39613754
- PMCID: PMC11607449
- DOI: 10.1038/s41467-024-54741-w
Paired analysis of host and pathogen genomes identifies determinants of human tuberculosis
Abstract
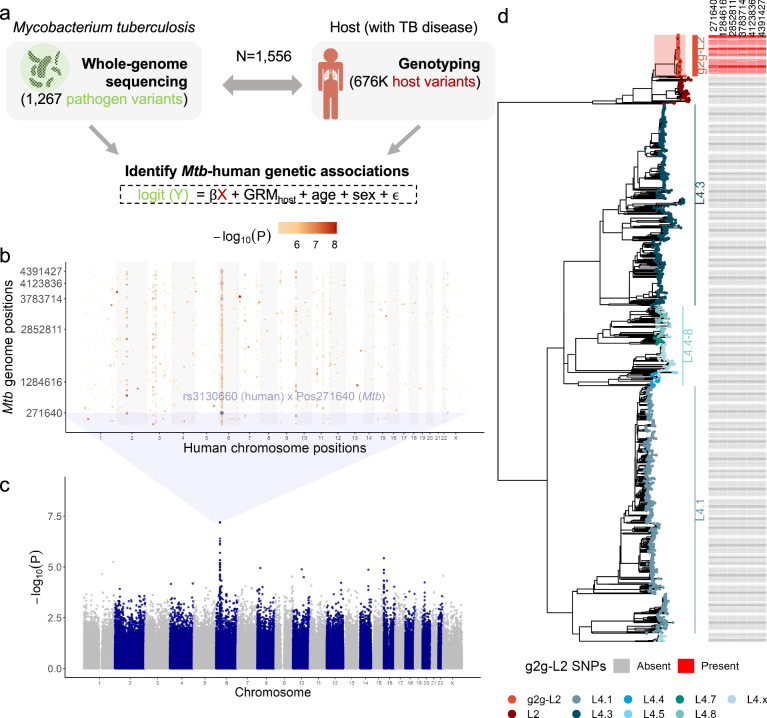
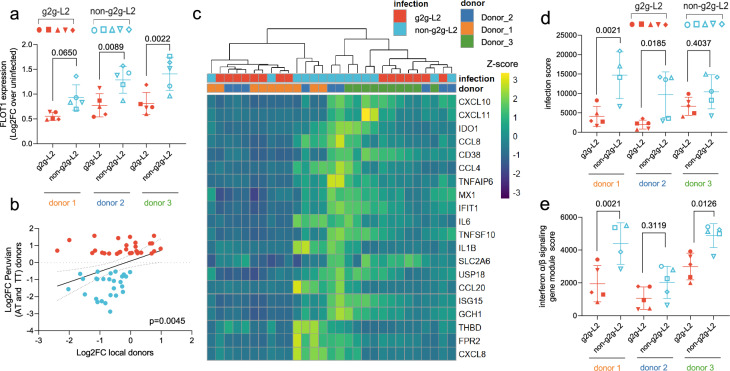
Infectious disease is the result of interactions between host and pathogen and can depend on genetic variations in both. We conduct a genome-to-genome study of paired human and Mycobacterium tuberculosis genomes from a cohort of 1556 tuberculosis patients in Lima, Peru. We identify an association between a human intronic variant (rs3130660, OR = 10.06, 95%CI: 4.87 - 20.77, P = 7.92 × 10-8) in the FLOT1 gene and a subclavaluee of Mtb Lineage 2. In a human macrophage infection model, we observe hosts with the rs3130660-A allele exhibited stronger interferon gene signatures. The interacting strains have altered redox states due to a thioredoxin reductase mutation. We investigate this association in a 2020 cohort of 699 patients recruited during the COVID-19 pandemic. While the prevalence of the interacting strain almost doubled between 2010 and 2020, its infection is not associated with rs3130660 in this recent cohort. These findings suggest a complex interplay among host, pathogen, and environmental factors in tuberculosis dynamics.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
-
- Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet.53, 1415–1424 (2021). - PubMed
-
- Organization, W. H. & Others. Global Tuberculosis Report 2018. 2018. Geneva: World Health Organizationhttps://iris.who.int/handle/10665/274453 (2019).
Publication types
MeSH terms
Substances
Associated data
- BioProject/PRJNA1039243
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
