

Characterizing the evolutionary dynamics of cancer proliferation in single-cell clones with SPRINTER
- PMID: 39614124
- PMCID: PMC11735394
- DOI: 10.1038/s41588-024-01989-z
Characterizing the evolutionary dynamics of cancer proliferation in single-cell clones with SPRINTER
Abstract
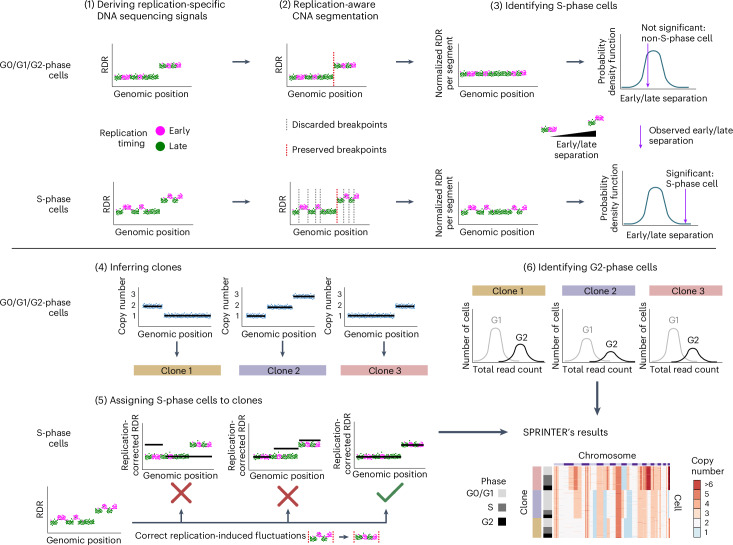
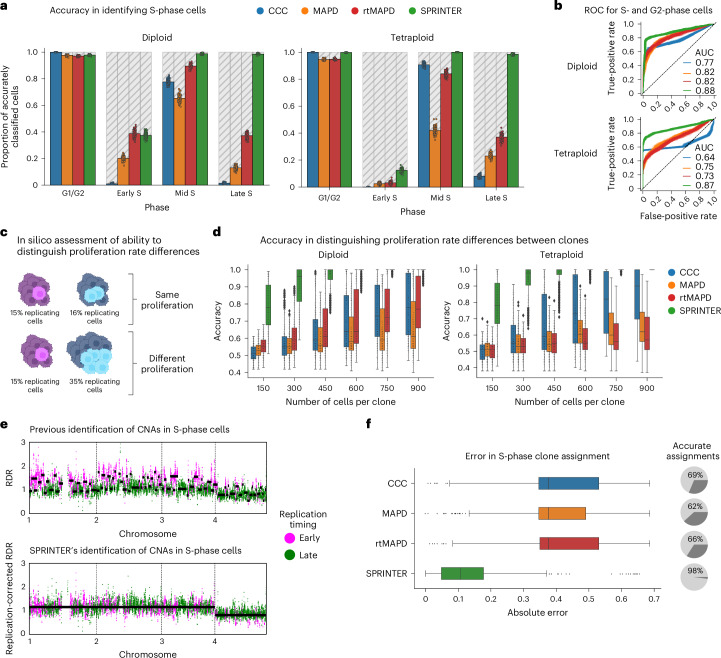
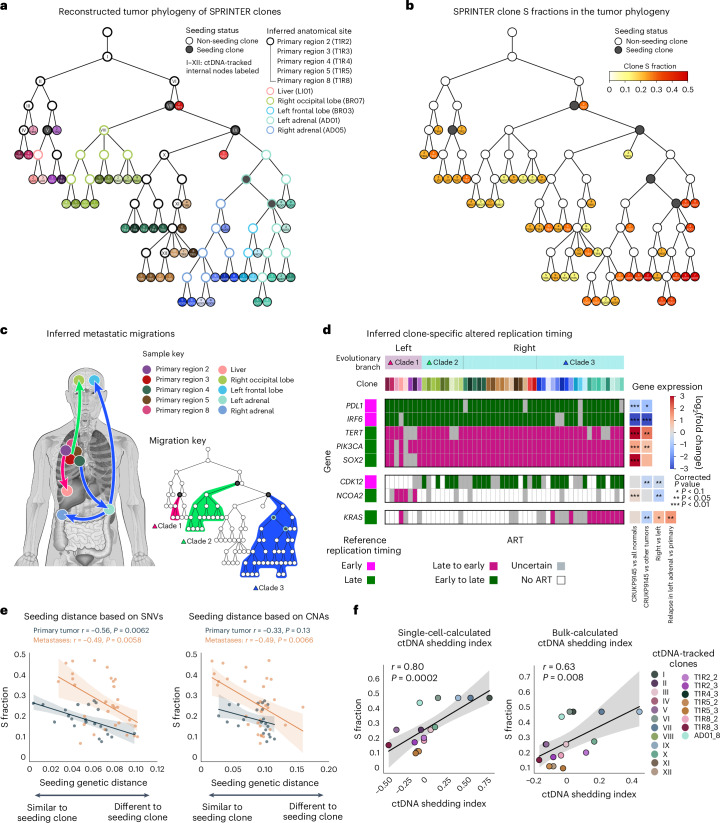
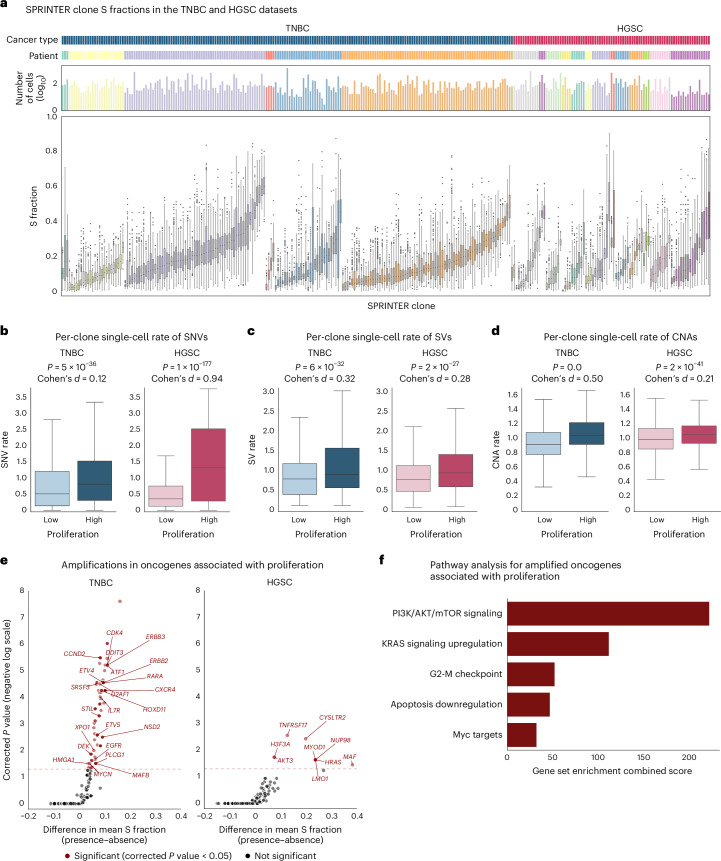
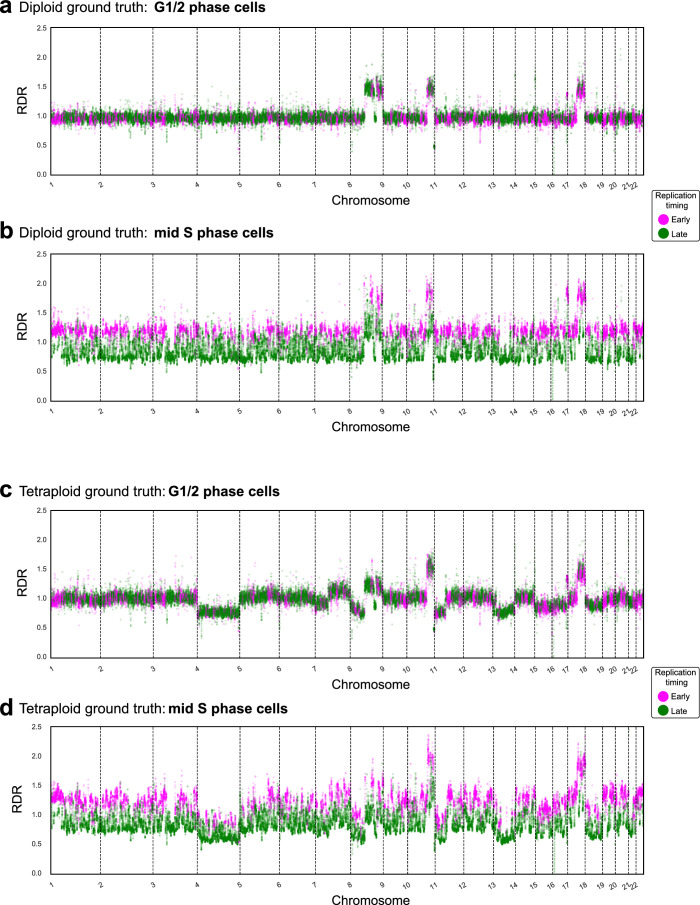
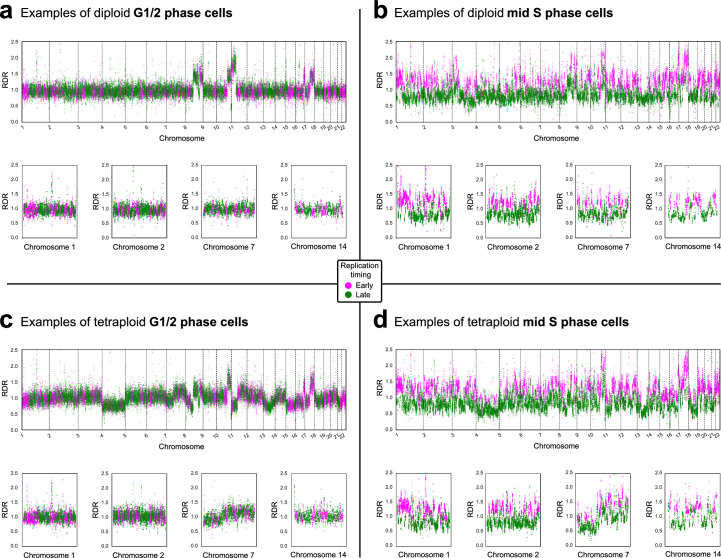
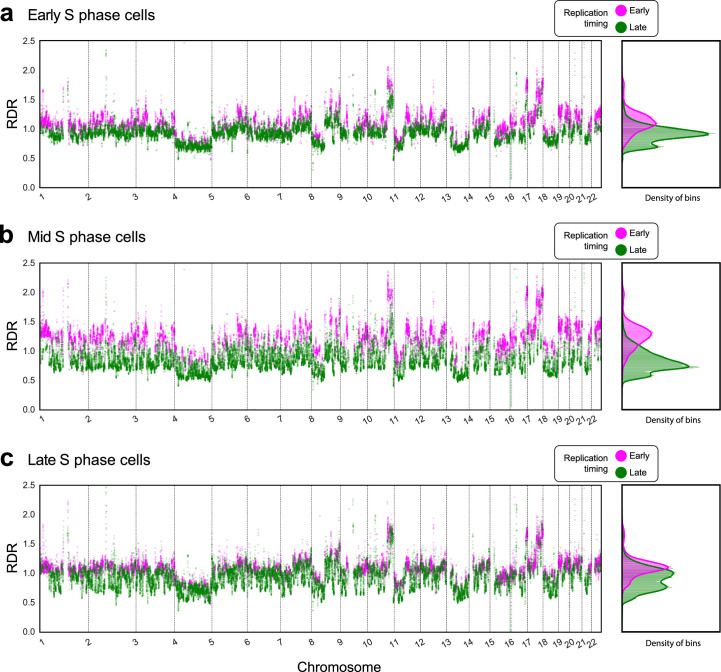
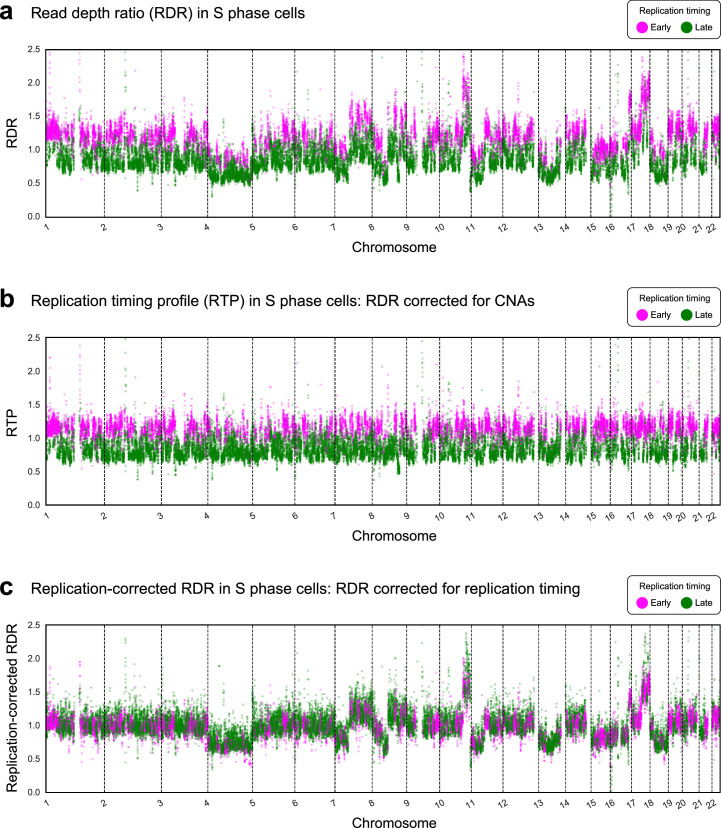
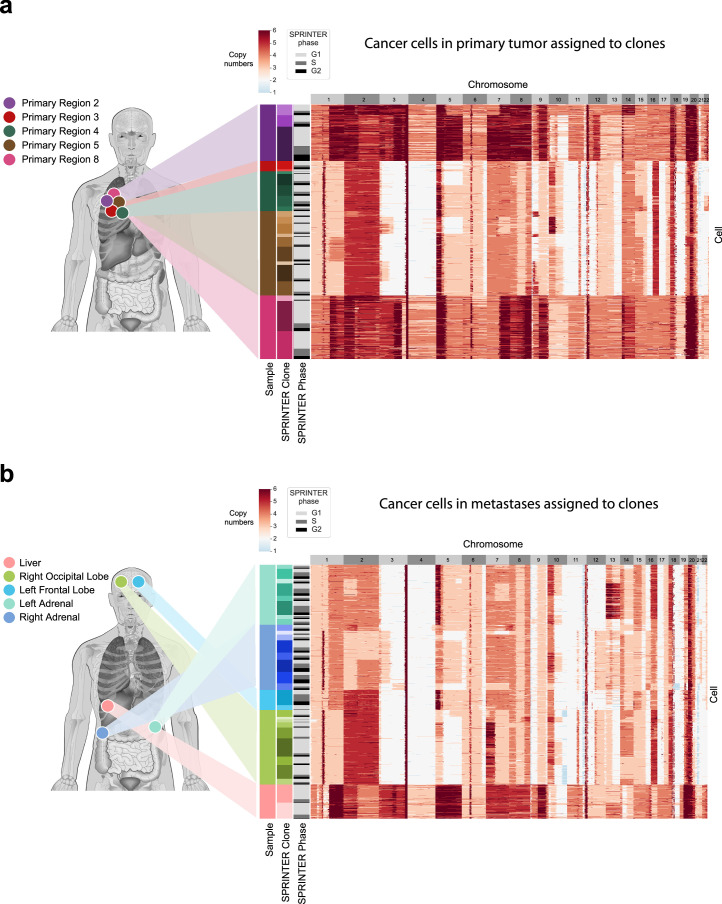
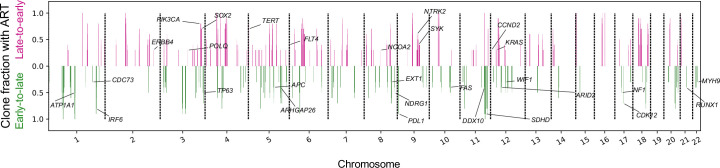
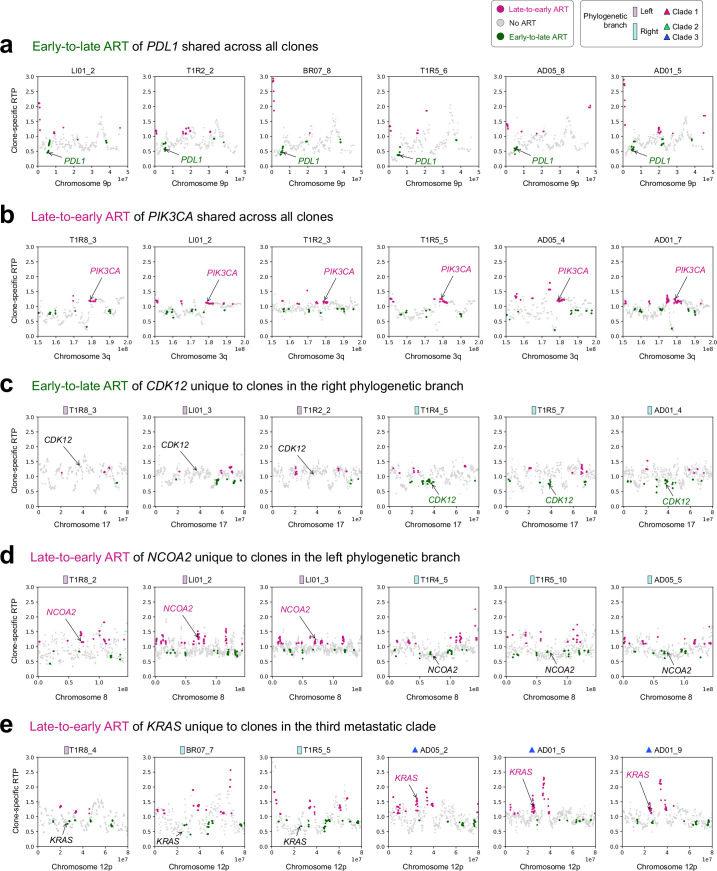
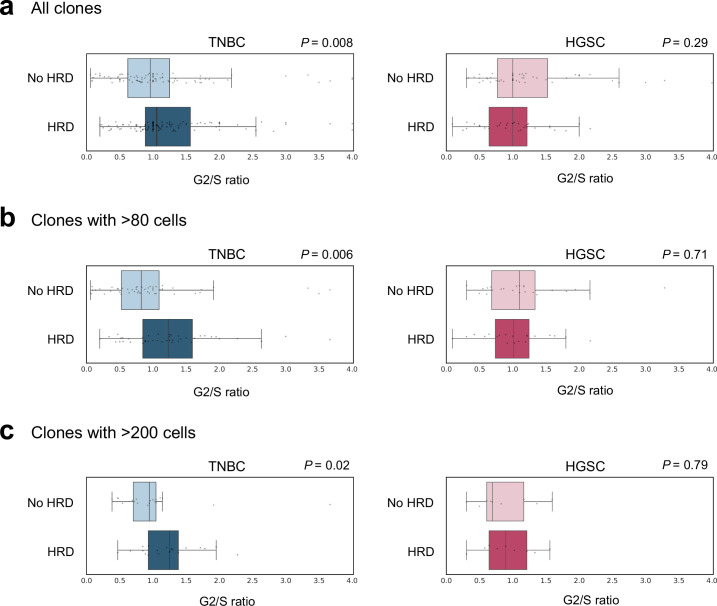
Proliferation is a key hallmark of cancer, but whether it differs between evolutionarily distinct clones co-existing within a tumor is unknown. We introduce the Single-cell Proliferation Rate Inference in Non-homogeneous Tumors through Evolutionary Routes (SPRINTER) algorithm that uses single-cell whole-genome DNA sequencing data to enable accurate identification and clone assignment of S- and G2-phase cells, as assessed by generating accurate ground truth data. Applied to a newly generated longitudinal, primary-metastasis-matched dataset of 14,994 non-small cell lung cancer cells, SPRINTER revealed widespread clone proliferation heterogeneity, orthogonally supported by Ki-67 staining, nuclei imaging and clinical imaging. We further demonstrated that high-proliferation clones have increased metastatic seeding potential, increased circulating tumor DNA shedding and clone-specific altered replication timing in proliferation- or metastasis-related genes associated with expression changes. Applied to previously generated datasets of 61,914 breast and ovarian cancer cells, SPRINTER revealed increased single-cell rates of different genomic variants and enrichment of proliferation-related gene amplifications in high-proliferation clones.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: A.M.F. is a co-inventor on a patent application to determine methods and systems for tumor monitoring (PCT/EP2022/077987). D.A.M. reports speaker fees from AstraZeneca and Takeda; consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol Myers Squibb and Eli Lilly and has received educational support from Takeda and Amgen. S.A. is a founder and shareholder of GenomeTherapeutics and scientific advisor to Sangamo Therapeutics, the Institute of Cancer Research, London, and the New York Genome Center, NY. N.M. has stock options in and has consulted for Achilles Therapeutics; holds a European patent in determining HLA LOH (PCT/GB2018/052004) and is a co-inventor to a patent to identifying responders to cancer treatment (PCT/GB2018/051912). M.J.-H. has received funding from CRUK, the National Institutes of Health (NIH) National Cancer Institute, International Association for the Study of Lung Cancer (IASLC) Foundation, Lung Cancer Research Foundation, Rosetrees Trust, UKI NETs and NIHR; has consulted for Astex Pharmaceutical and Achilles Therapeutics; is a member of the Achilles Therapeutics Scientific Advisory Board and Steering Committee and has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, Bristol Myers Squibb and Genentech. M.J.-H. is listed as a co-inventor on a European patent application relating to methods to detect lung cancer (PCT/US2017/028013); this patent has been licensed to commercial entities, and, under terms of employment, M.J.-H. is due a share of any revenue generated from such license(s) and is also listed as a co-inventor on the GB priority patent application (GB2400424.4) with title—Treatment and Prevention of Lung Cancer. C.S. acknowledges grants from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx—collaboration in minimal residual disease sequencing technologies), Ono Pharmaceutical and Personalis. He is the chief investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also the co-chief investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board (SAB). He receives consultant fees from Achilles Therapeutics (also a SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, the China Innovation Centre of Roche (CICoR), formerly Roche Innovation Centre—Shanghai, Metabomed (until July 2022), Relay Therapeutics SAB member, Saga Diagnostics SAB member and the Sarah Cannon Research Institute. C.S. has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL; currently has stock options in Epic Bioscience, Bicycle Therapeutics and Relay Therapeutics; and has stock options and is cofounder of Achilles Therapeutics. C.S. declares a patent application for methods to lung cancer (PCT/US2017/028013), targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), methods for lung cancer detection (US20190106751A1), identifying patients who respond to cancer treatment (PCT/GB2018/051912), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), methods and systems for tumor monitoring (PCT/EP2022/077987). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumor recurrence. This patent has been licensed to a commercial entity, and, under their terms of employment, C.S. is due a revenue share of any revenue generated from such license(s). The other authors declare no competing interests.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
