

Effective data visualization strategies in untargeted metabolomics
- PMID: 39620439
- PMCID: PMC11610048
- DOI: 10.1039/d4np00039k
Effective data visualization strategies in untargeted metabolomics
Abstract
Covering: 2014 to 2023 for metabolomics, 2002 to 2023 for information visualizationLC-MS/MS-based untargeted metabolomics is a rapidly developing research field spawning increasing numbers of computational metabolomics tools assisting researchers with their complex data processing, analysis, and interpretation tasks. In this article, we review the entire untargeted metabolomics workflow from the perspective of information visualization, visual analytics and visual data integration. Data visualization is a crucial step at every stage of the metabolomics workflow, where it provides core components of data inspection, evaluation, and sharing capabilities. However, due to the large number of available data analysis tools and corresponding visualization components, it is hard for both users and developers to get an overview of what is already available and which tools are suitable for their analysis. In addition, there is little cross-pollination between the fields of data visualization and metabolomics, leaving visual tools to be designed in a secondary and mostly ad hoc fashion. With this review, we aim to bridge the gap between the fields of untargeted metabolomics and data visualization. First, we introduce data visualization to the untargeted metabolomics field as a topic worthy of its own dedicated research, and provide a primer on cutting-edge visualization research into data visualization for both researchers as well as developers active in metabolomics. We extend this primer with a discussion of best practices for data visualization as they have emerged from data visualization studies. Second, we provide a practical roadmap to the visual tool landscape and its use within the untargeted metabolomics field. Here, for several computational analysis stages within the untargeted metabolomics workflow, we provide an overview of commonly used visual strategies with practical examples. In this context, we will also outline promising areas for further research and development. We end the review with a set of recommendations for developers and users on how to make the best use of visualizations for more effective and transparent communication of results.
Conflict of interest statement
J. J. J. van der Hooft is currently a member of the Scientific Advisory Board of NAICONS Srl., Milano, Italy, and is consulting for Corteva Agriscience, Indianapolis, IN, USA. All other authors declare no conflict of interest.
Figures














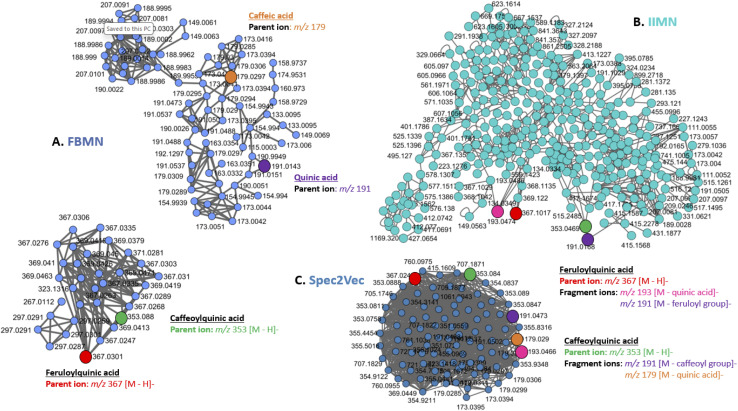








References
-
- Munzner T., Visualization Analysis and Design, CRC Press, 2014
-
- Matejka J. and Fitzmaurice G., Proceedings of the 2017 CHI Conference on Human Factors in Computing Systems, 2017
-
- Murray L. L. Wilson J. G. Decis. Sci. J. Innov. Educ. 2021;19:157–166.
-
- Andrienko N., Andrienko G., Fuchs G., Slingsby A., Turkay C. and Wrobel S., in Visual Analytics for Investigating and Processing Data, Springer International Publishing, 2020, pp. 151–180
-
- Liu S. Cui W. Wu Y. Liu M. Vis. Comput. 2014;30:1373–1393. doi: 10.1007/s00371-013-0892-3. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous