

Noninvasive Multicancer Detection Using DNA Hypomethylation of LINE-1 Retrotransposons
- PMID: 39620930
- PMCID: PMC11959274
- DOI: 10.1158/1078-0432.CCR-24-2669
Noninvasive Multicancer Detection Using DNA Hypomethylation of LINE-1 Retrotransposons
Abstract
Purpose: The detection of ctDNA, which allows noninvasive tumor molecular profiling and disease follow-up, promises optimal and individualized management of patients with cancer. However, detecting small fractions of tumor DNA released when the tumor burden is reduced remains a challenge.
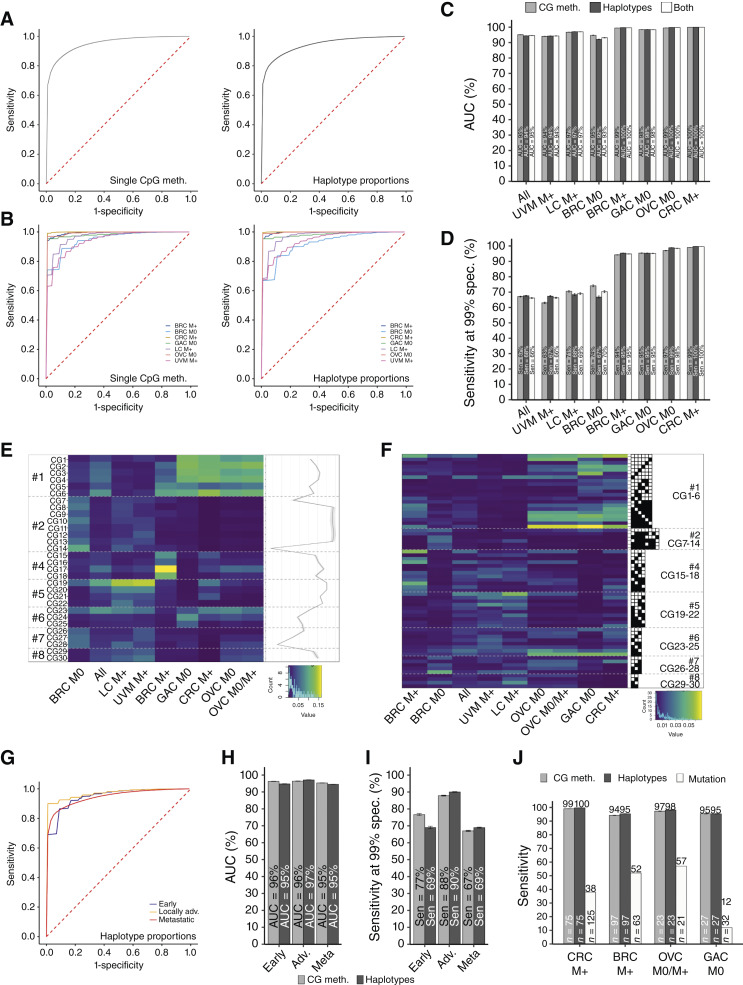
Experimental design: We implemented a new, highly sensitive strategy to detect bp resolution methylation patterns from plasma DNA and assessed the potential of hypomethylation of long interspersed nuclear element-1 retrotransposons as a noninvasive multicancer detection biomarker. The Detection of Long Interspersed Nuclear Element Altered Methylation ON plasma DNA method targets 30 to 40,000 young long interspersed nuclear element-1 retrotransposons scattered throughout the genome, covering about 100,000 CpG sites and is based on a reference-free analysis pipeline.
Results: Resulting machine learning-based classifiers showed powerful correct classification rates discriminating healthy and tumor plasmas from six types of cancers (colorectal, breast, lung, ovarian, and gastric cancers and uveal melanoma, including localized stages) in two independent cohorts (AUC = 88%-100%, N = 747). The Detection of Long Interspersed Nuclear Element Altered Methylation ON plasma DNA method can also be used to perform copy number alteration analysis that improves cancer detection.
Conclusions: This should lead to the development of more efficient noninvasive diagnostic tests adapted to all patients with cancer, based on the universality of these factors. See related commentary by Szymanski et al., p. 1179.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
M. Michel reports a patent for PCT/EP2023/074092—Sensitive and Specific Determination of DNA Methylation Profiles pending. M. Heidary reports grants from the European Research Council (ERC-StG EpiDetect), the Ligue contre le cancer (RS17-75-75), the prematuration program of the Centre National pour la Recherche Scientifique, the SiRIC 2 Curie program (INCa-DGOS-Inserm_12554), the DEEP Strive funding (LABEX DEEP 11-LBX0044), ANR-10-EQPX-03 (Equipex), ANR-10-INBS-09-08 (France Genomique Consortium), and ANR-19-P3IA-0001 (PRAIRIE 3IA Institute) during the conduct of the study, as well as a patent for “PCT/EP2023/074092—Sensitive and Specific Determination of DNA Methylation Profiles—Inventors: Proudhon, Charlotte; Azencott, Chloé-Agathe; Michel, Marc; Heidary, Maryam” pending. M. Kamal reports personal fees from Roche outside the submitted work. C. Le Tourneau reports personal fees from MSD, Bristol Myers Squibb, Merck, AstraZeneca, Celgene, Seattle Genetics, Roche, Novartis, Rakuten, Nanobiotix, and GSK outside the submitted work. M.-H. Stern reports a patent for US20190256921A1 pending, a patent for ES2978017T3 issued, a patent for PCT/EP2019/056445 issued, and a patent for PCT/EP2023/057543 issued. C.-A. Azencott reports grants from Agence Nationale de la Recherche during the conduct of the study, as well as grants from Janssen Research & Development outside the submitted work; in addition, C.-A. Azencott reports a patent for WO2024047250—Sensitive and Specific Determination of DNA Methylation Profiles pending. C. Proudhon reports grants from the European Research Council, the Ligue contre le cancer, the French National Center for Scientific Research, grants from SiRIC 2 Curie program, and the DEEP Strive funding during the conduct of the study, as well as a patent for PCT/EP2023/074092—Sensitive and Specific Determination of DNA Methylation Profiles—Inventors: Proudhon, Charlotte; Azencott, Chloé-Agathe; Michel, Marc; Heidary, Maryam pending. No disclosures were reported by the other authors.
Figures




References
-
- Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. . Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133. - PubMed
-
- Bidard F-C, Weigelt B, Reis-Filho JS. Going with the flow: from circulating tumor cells to DNA. Sci Transl Med 2013;5:207ps14. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
