

FABIO: TWAS fine-mapping to prioritize causal genes for binary traits
- PMID: 39621803
- PMCID: PMC11649093
- DOI: 10.1371/journal.pgen.1011503
FABIO: TWAS fine-mapping to prioritize causal genes for binary traits
Abstract
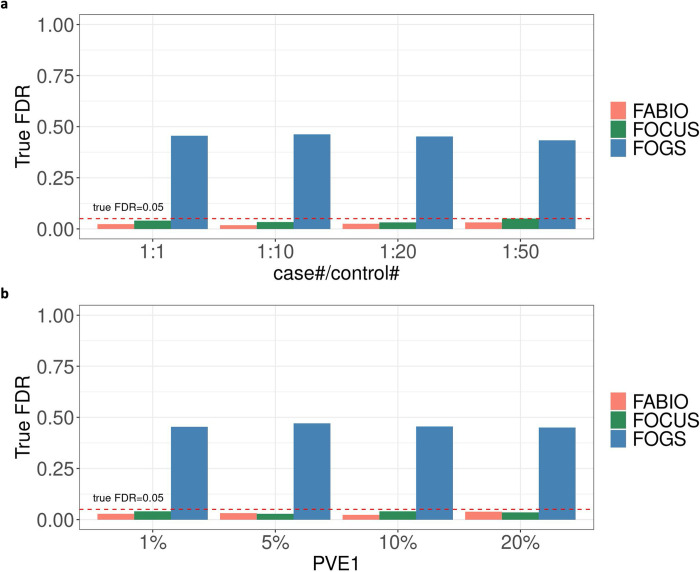
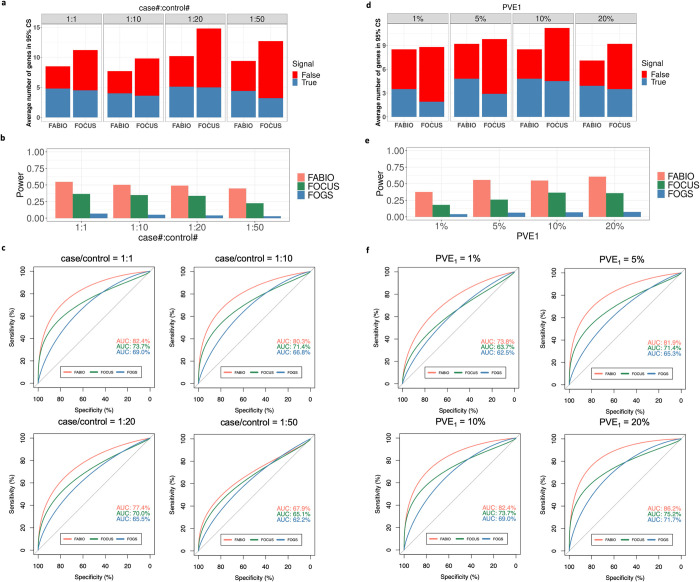
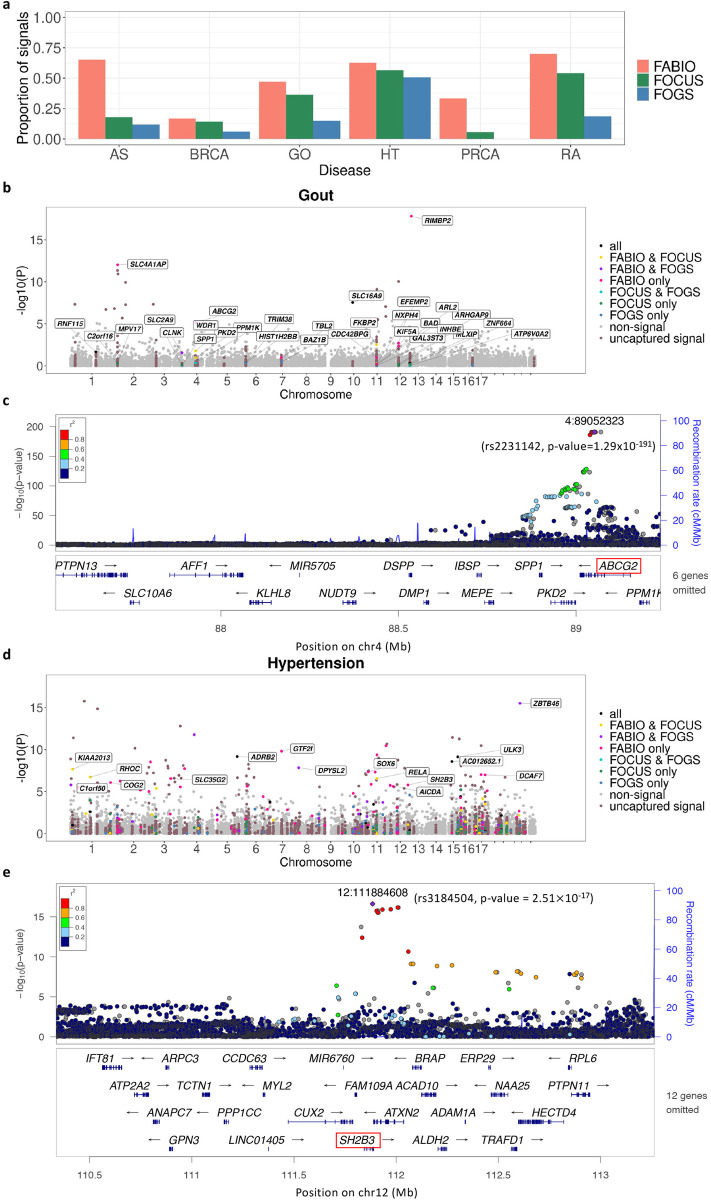
Transcriptome-wide association studies (TWAS) have emerged as a powerful tool for identifying gene-trait associations by integrating gene expression mapping studies with genome-wide association studies (GWAS). While most existing TWAS approaches focus on marginal analyses through examining one gene at a time, recent developments in TWAS fine-mapping methods enable the joint modeling of multiple genes to refine the identification of potentially causal ones. However, these fine-mapping methods have primarily focused on modeling quantitative traits and examining local genomic regions, leading to potentially suboptimal performance. Here, we present FABIO, a TWAS fine-mapping method specifically designed for binary traits that is capable of modeling all genes jointly on an entire chromosome. FABIO employs a probit model to directly link the genetically regulated expression (GReX) of genes to binary outcomes while taking into account the GReX correlation among all genes residing on a chromosome. As a result, FABIO effectively controls false discoveries while offering substantial power gains over existing TWAS fine-mapping approaches. We performed extensive simulations to evaluate the performance of FABIO and applied it for in-depth analyses of six binary disease traits in the UK Biobank. In the real datasets, FABIO significantly reduced the size of the causal gene sets by 27.9%-36.9% over existing approaches across traits. Leveraging its improved power, FABIO successfully prioritized multiple potentially causal genes associated with the diseases, including GATA3 for asthma, ABCG2 for gout, and SH2B3 for hypertension. Overall, FABIO represents an effective tool for TWAS fine-mapping of disease traits.
Copyright: © 2024 Zhang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: L.C.T has received support from Galderma and Janssen.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
