

Borrelia PeptideAtlas: A proteome resource of common Borrelia burgdorferi isolates for Lyme research
- PMID: 39622905
- PMCID: PMC11612207
- DOI: 10.1038/s41597-024-04047-9
Borrelia PeptideAtlas: A proteome resource of common Borrelia burgdorferi isolates for Lyme research
Abstract
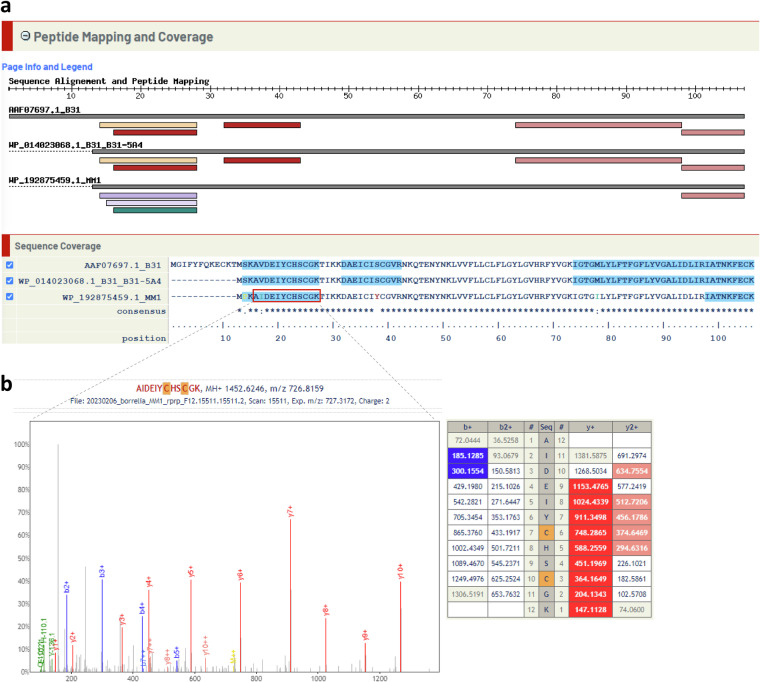
Lyme disease is caused by an infection with the spirochete Borrelia burgdorferi, and is the most common vector-borne disease in North America. B. burgdorferi isolates harbor extensive genomic and proteomic variability and further comparison of isolates is key to understanding the infectivity of the spirochetes and biological impacts of identified sequence variants. Here, we applied both transcriptome analysis and mass spectrometry-based proteomics to assemble peptide datasets of B. burgdorferi laboratory isolates B31, MM1, and the infective isolate B31-5A4, to provide a publicly available Borrelia PeptideAtlas. Included are total proteome, secretome, and membrane proteome identifications of the individual isolates. Proteomic data collected from 35 different experiment datasets, totaling 386 mass spectrometry runs, have identified 81,967 distinct peptides, which map to 1,113 proteins. The Borrelia PeptideAtlas covers 86% of the total B31 proteome of 1,291 protein sequences. The Borrelia PeptideAtlas is an extensible comprehensive peptide repository with proteomic information from B. burgdorferi isolates useful for Lyme disease research.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
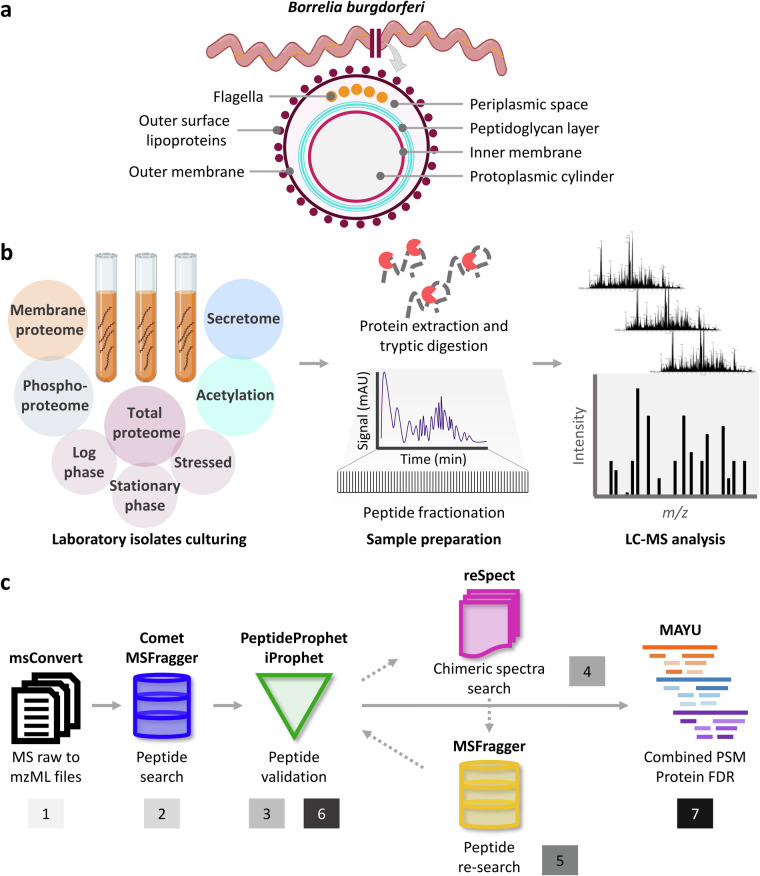
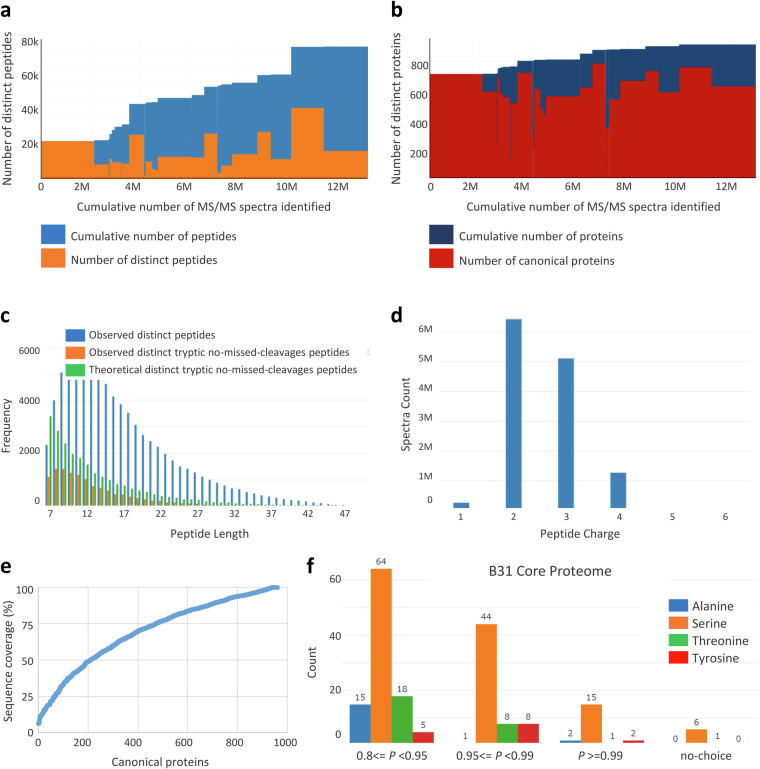

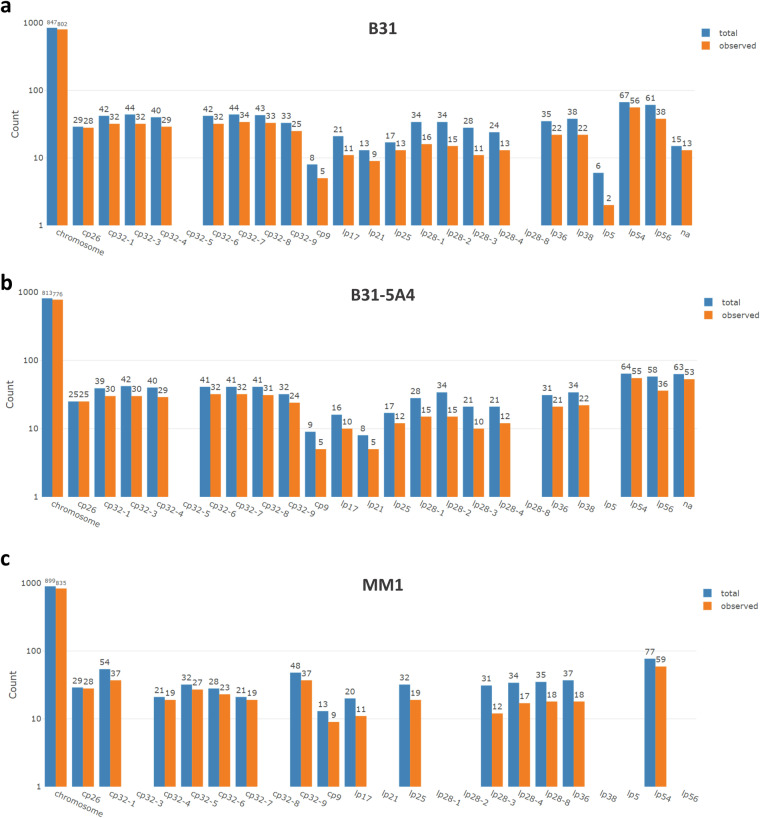
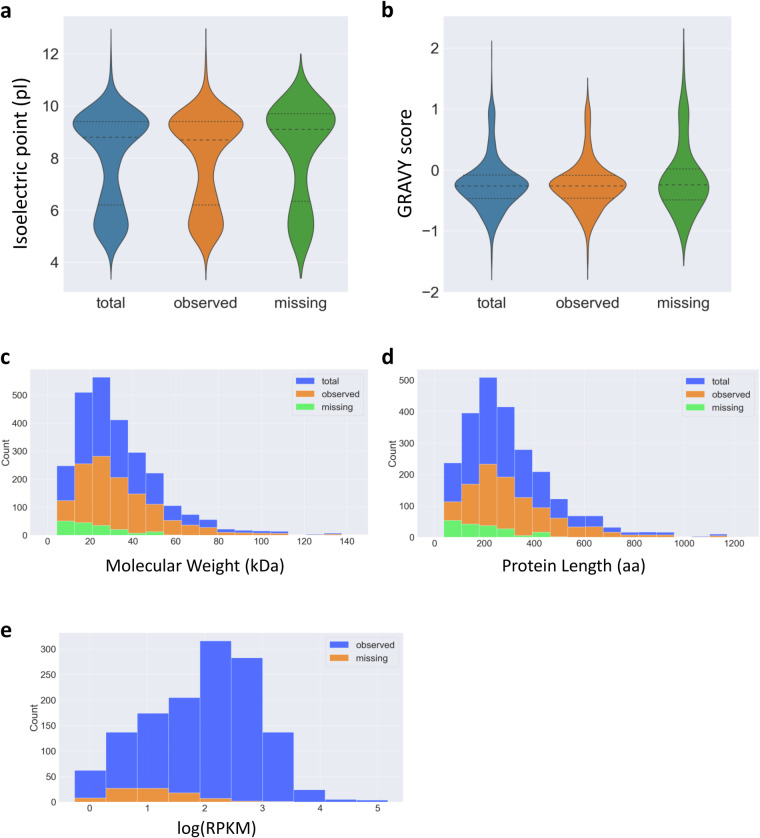
Update of
-
Borrelia PeptideAtlas: A proteome resource of common Borrelia burgdorferi isolates for Lyme research.bioRxiv [Preprint]. 2023 Jun 16:2023.06.16.545244. doi: 10.1101/2023.06.16.545244. bioRxiv. 2023. Update in: Sci Data. 2024 Dec 2;11(1):1313. doi: 10.1038/s41597-024-04047-9. PMID: 37398146 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- 1920268/National Science Foundation (NSF)
- R01AI029735/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R21 AI142302/AI/NIAID NIH HHS/United States
- S10 OD026936/OD/NIH HHS/United States
- R21AI133335/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 GM087221/GM/NIGMS NIH HHS/United States
- R21AI142302/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 AI029735/AI/NIAID NIH HHS/United States
- R21 AI133335/AI/NIAID NIH HHS/United States
- R01GM087221/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
