

RNA-Puzzles Round V: blind predictions of 23 RNA structures
- PMID: 39623050
- PMCID: PMC11810798
- DOI: 10.1038/s41592-024-02543-9
RNA-Puzzles Round V: blind predictions of 23 RNA structures
Abstract
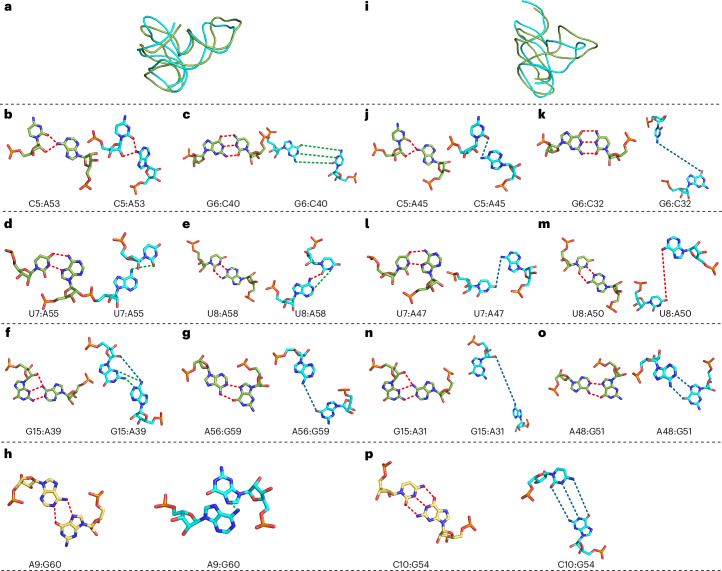
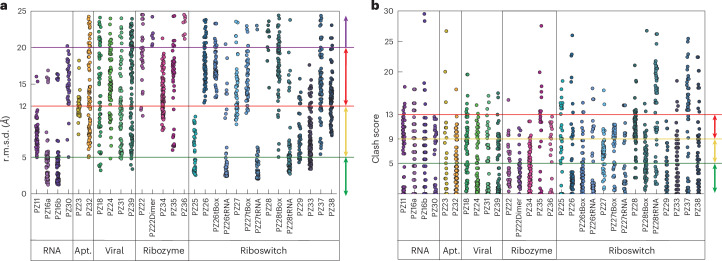
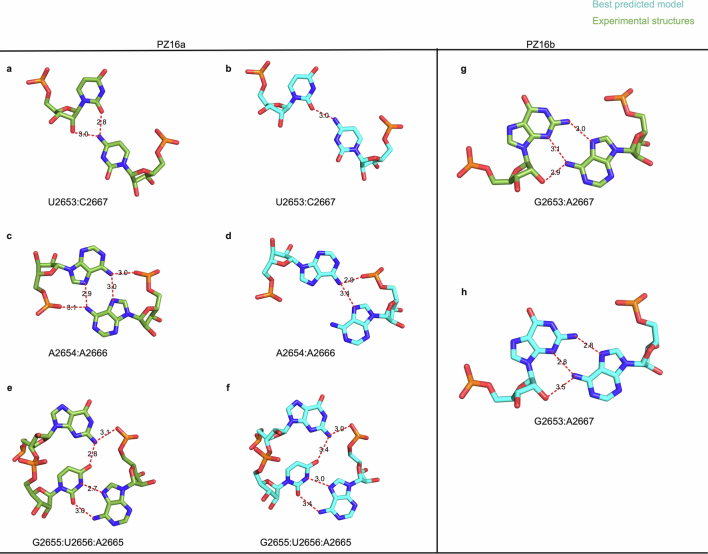
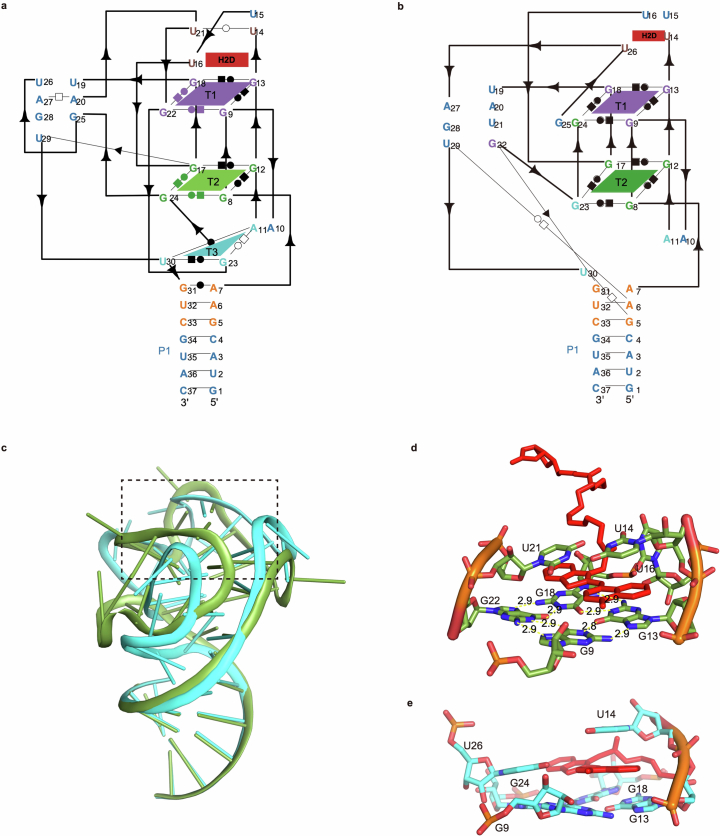
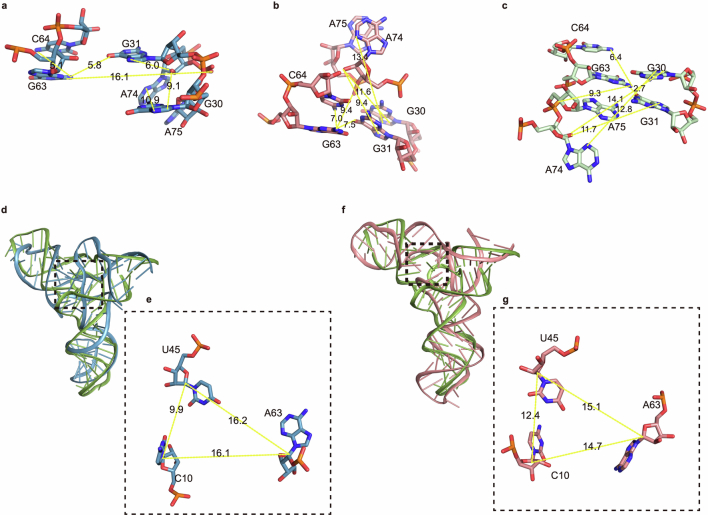
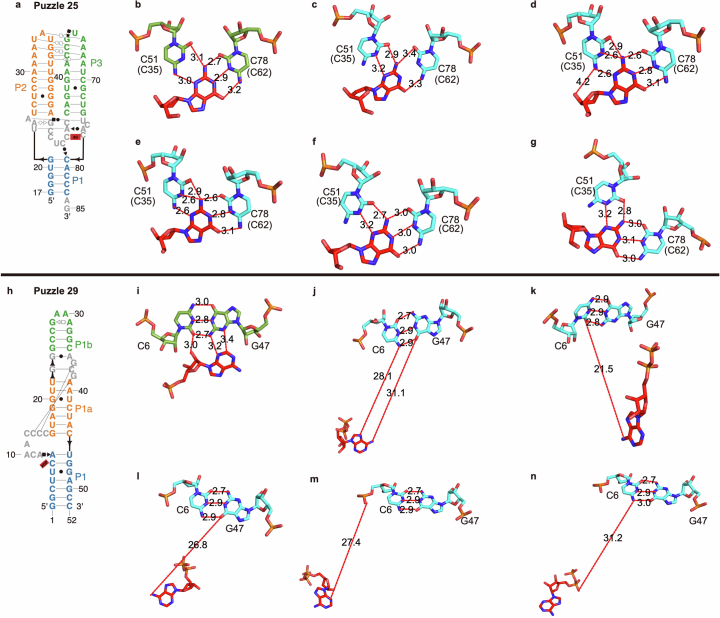
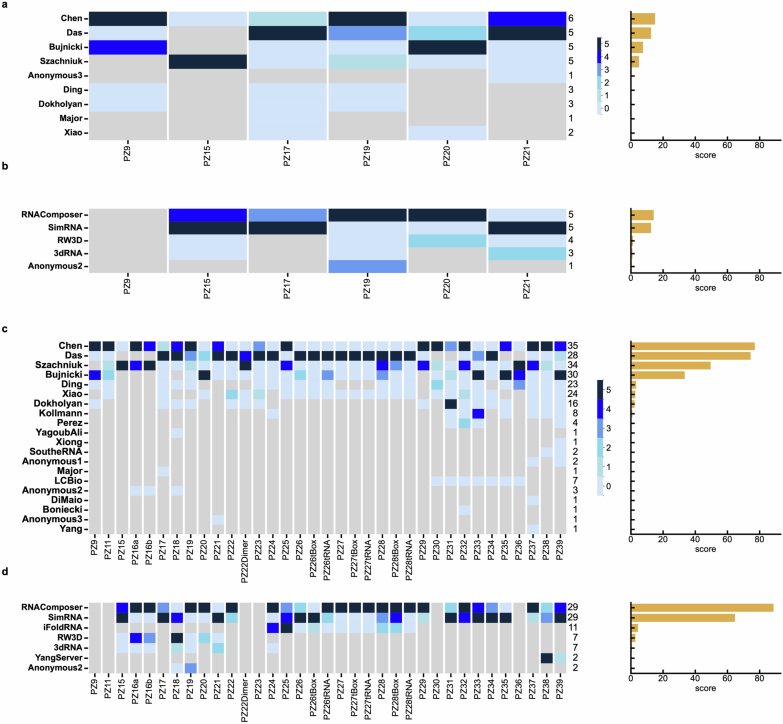
RNA-Puzzles is a collective endeavor dedicated to the advancement and improvement of RNA three-dimensional structure prediction. With agreement from structural biologists, RNA structures are predicted by modeling groups before publication of the experimental structures. We report a large-scale set of predictions by 18 groups for 23 RNA-Puzzles: 4 RNA elements, 2 Aptamers, 4 Viral elements, 5 Ribozymes and 8 Riboswitches. We describe automatic assessment protocols for comparisons between prediction and experiment. Our analyses reveal some critical steps to be overcome to achieve good accuracy in modeling RNA structures: identification of helix-forming pairs and of non-Watson-Crick modules, correct coaxial stacking between helices and avoidance of entanglements. Three of the top four modeling groups in this round also ranked among the top four in the CASP15 contest.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
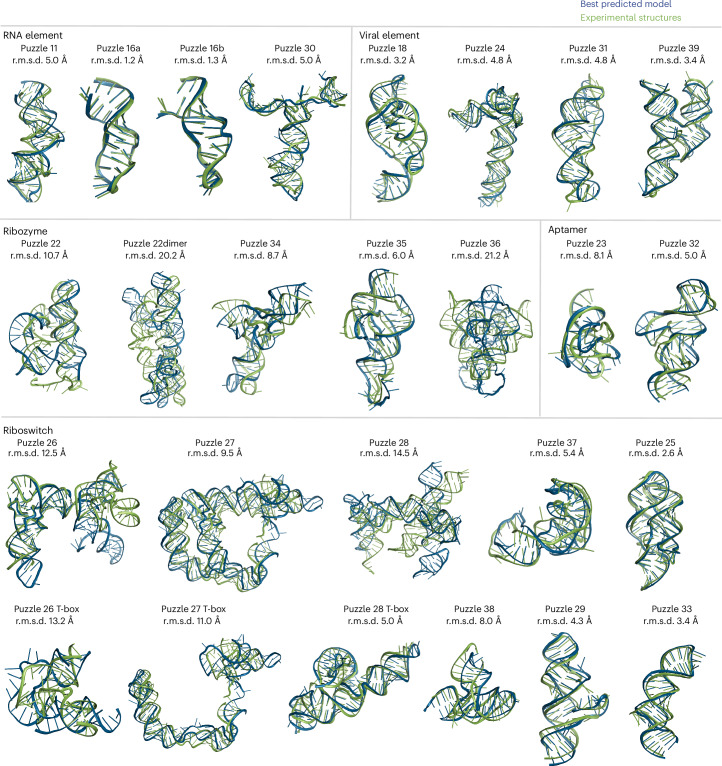
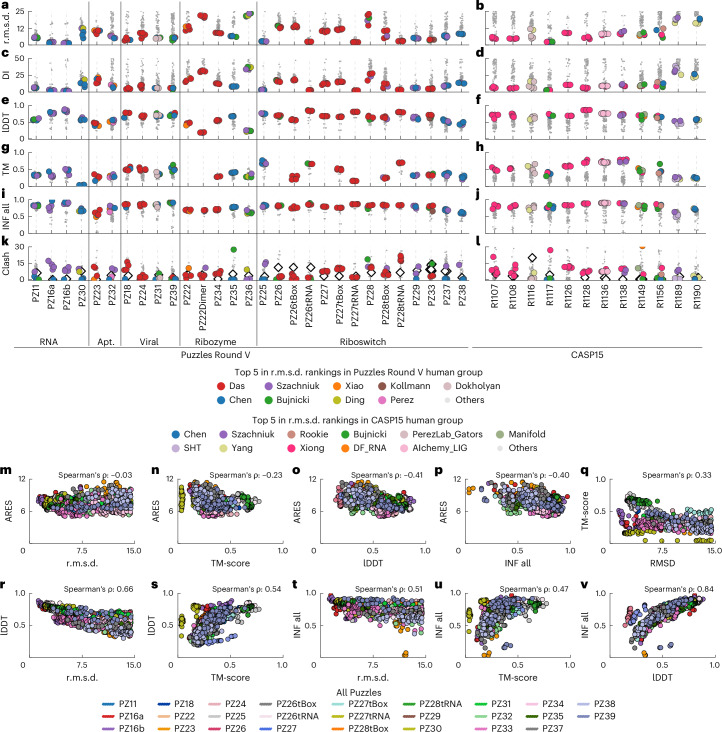
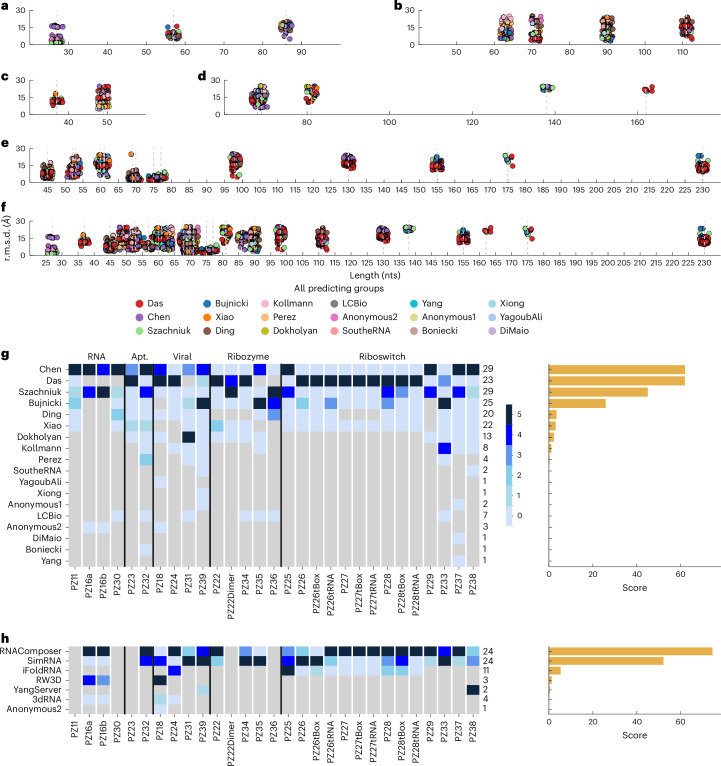
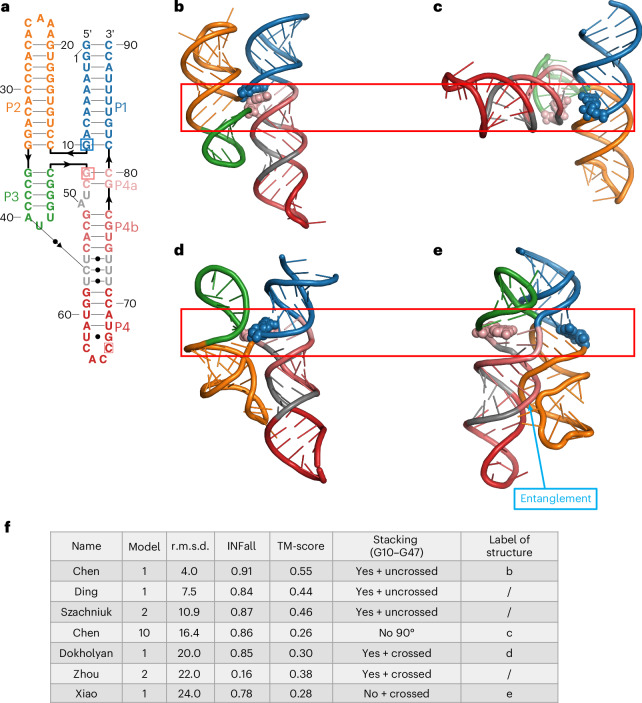
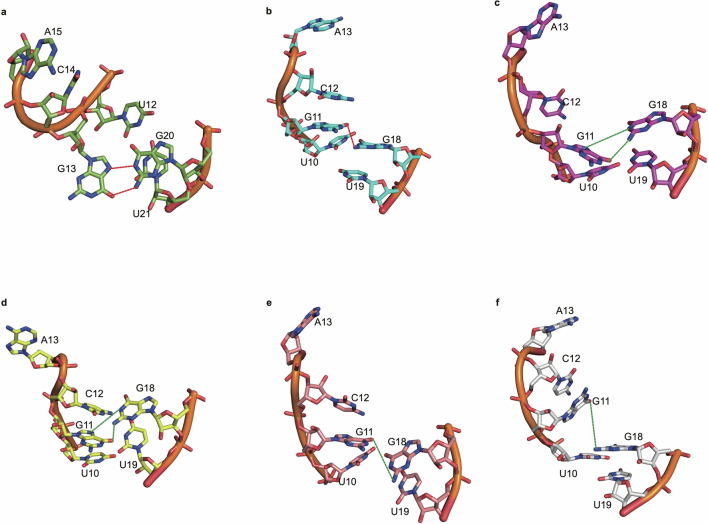
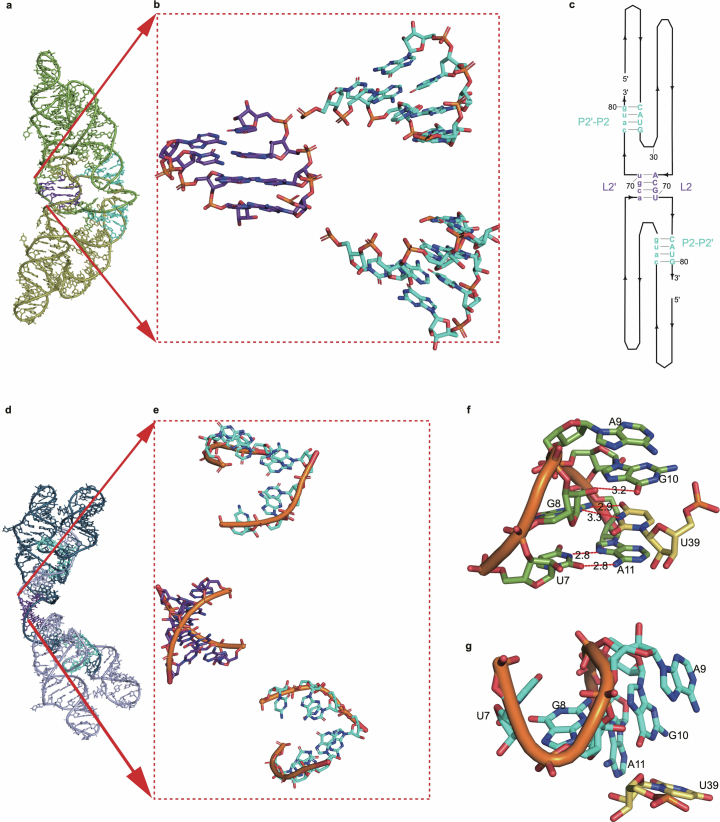
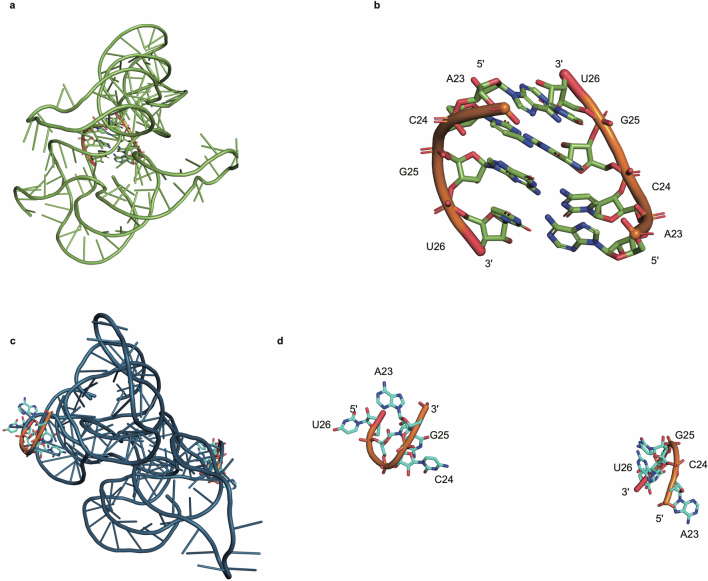
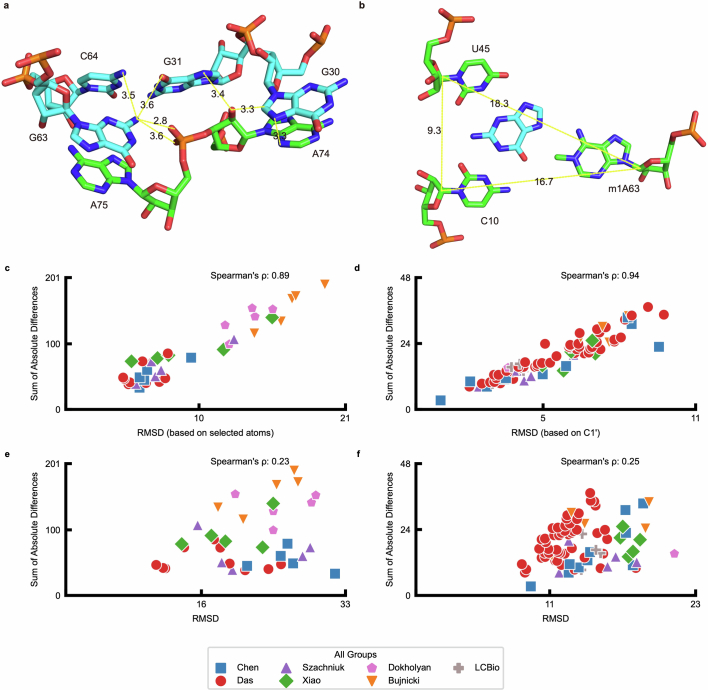
References
-
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A32, 922–923 (1976).
MeSH terms
Substances
Grants and funding
- T32 GM066706/GM/NIGMS NIH HHS/United States
- NSFC T2225007/National Natural Science Foundation of China (National Science Foundation of China)
- R35 GM134919/GM/NIGMS NIH HHS/United States
- R35 GM149336/GM/NIGMS NIH HHS/United States
- R35GM145409/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- ZIA DK075136/ImNIH/Intramural NIH HHS/United States
- R35 GM145409/GM/NIGMS NIH HHS/United States
- 32270707/National Natural Science Foundation of China (National Science Foundation of China)
- R35 GM122579/GM/NIGMS NIH HHS/United States
- R35 GM134864/GM/NIGMS NIH HHS/United States
- R01 AI133348/AI/NIAID NIH HHS/United States
- T32 grant GM066706/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- P20GM121342/Foundation for the National Institutes of Health (Foundation for the National Institutes of Health, Inc.)
- F32 GM112294/GM/NIGMS NIH HHS/United States
- R21 CA219847/CA/NCI NIH HHS/United States
- 32171191/National Natural Science Foundation of China (National Science Foundation of China)
- P20 GM121342/GM/NIGMS NIH HHS/United States
- R35 GM152029/GM/NIGMS NIH HHS/United States
- R01 GM073850/GM/NIGMS NIH HHS/United States
