

Single-cell RNA sequencing of peripheral blood links cell-type-specific regulation of splicing to autoimmune and inflammatory diseases
- PMID: 39627432
- PMCID: PMC11631754
- DOI: 10.1038/s41588-024-02019-8
Single-cell RNA sequencing of peripheral blood links cell-type-specific regulation of splicing to autoimmune and inflammatory diseases
Abstract
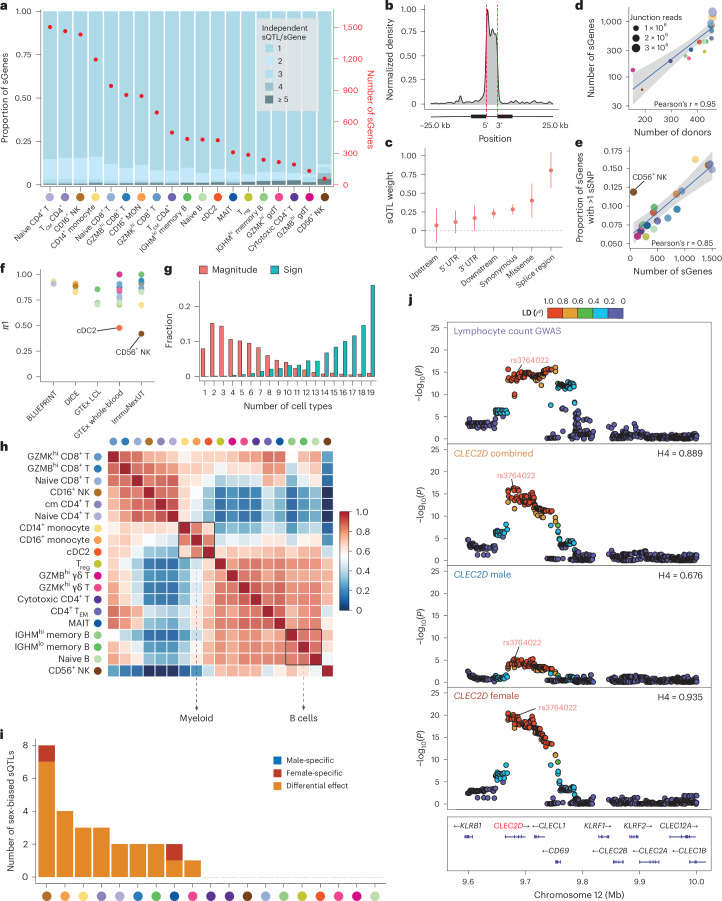
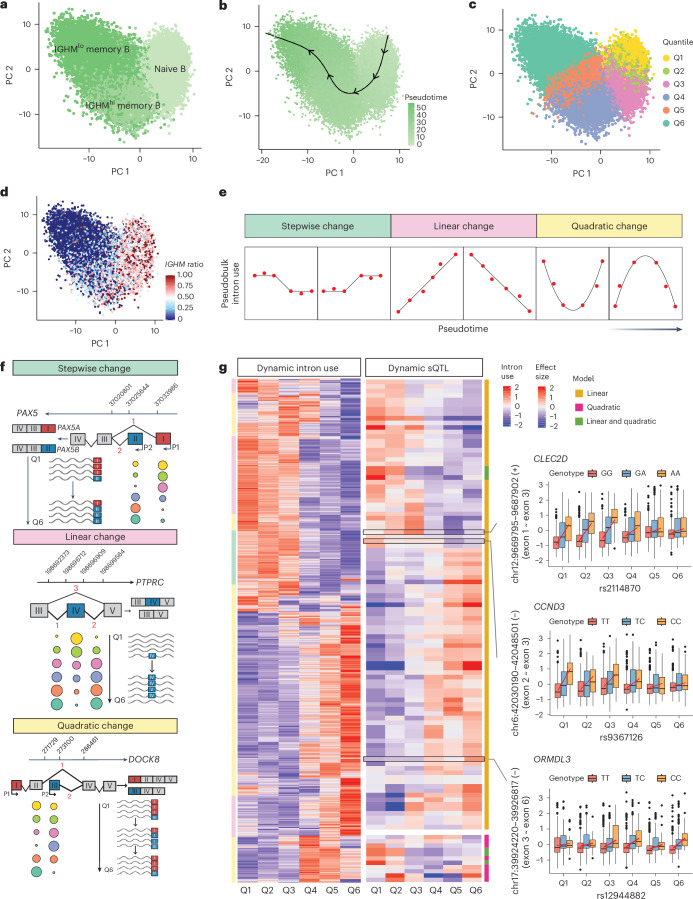
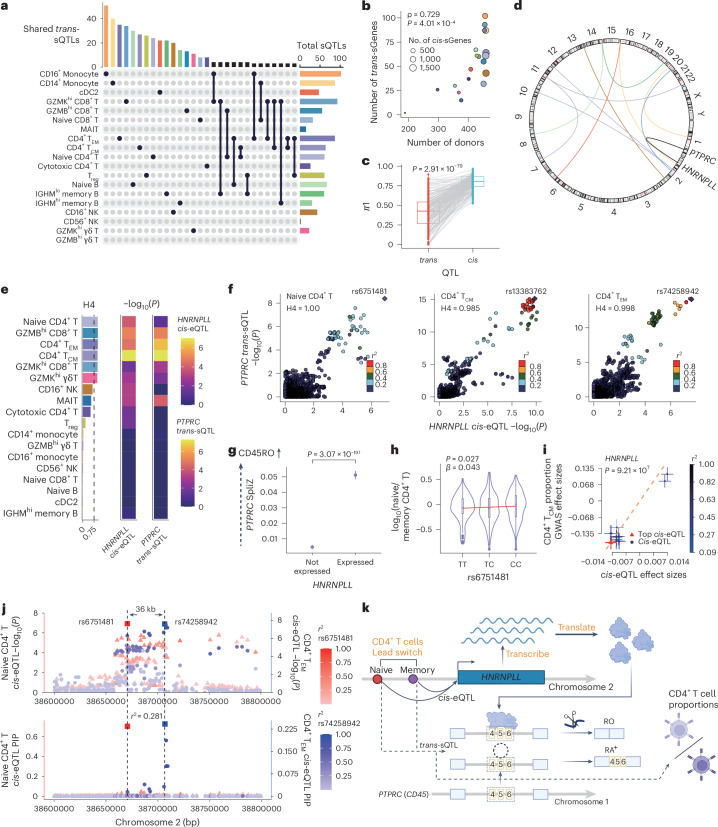
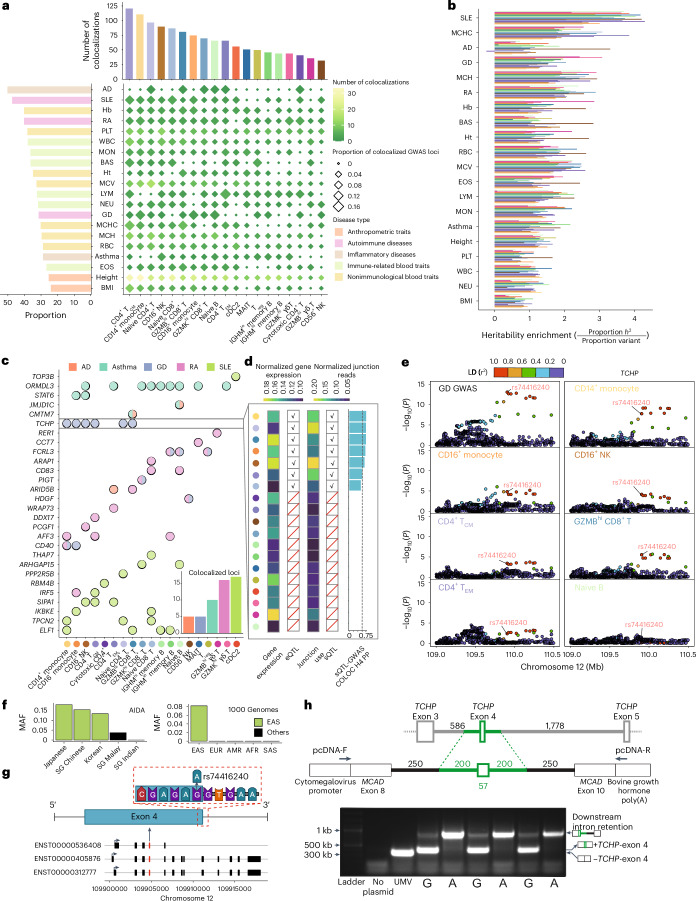
Alternative splicing contributes to complex traits, but whether this differs in trait-relevant cell types across diverse genetic ancestries is unclear. Here we describe cell-type-specific, sex-biased and ancestry-biased alternative splicing in ~1 M peripheral blood mononuclear cells from 474 healthy donors from the Asian Immune Diversity Atlas. We identify widespread sex-biased and ancestry-biased differential splicing, most of which is cell-type-specific. We identify 11,577 independent cis-splicing quantitative trait loci (sQTLs), 607 trans-sGenes and 107 dynamic sQTLs. Colocalization between cis-eQTLs and trans-sQTLs revealed a cell-type-specific regulatory relationship between HNRNPLL and PTPRC. We observed an enrichment of cis-sQTL effects in autoimmune and inflammatory disease heritability. Specifically, we functionally validated an Asian-specific sQTL disrupting the 5' splice site of TCHP exon 4 that putatively modulates the risk of Graves' disease in East Asian populations. Our work highlights the impact of ancestral diversity on splicing and provides a roadmap to dissect its role in complex diseases at single-cell resolution.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: X.J. is an employee of BGI Research. Y. Tong is undertaking a PhD scholarship partially supported by BGI Research. The other authors declare no competing interests.
Figures












References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
