

Using multiplexed functional data to reduce variant classification inequities in underrepresented populations
- PMID: 39627863
- PMCID: PMC11616159
- DOI: 10.1186/s13073-024-01392-7
Using multiplexed functional data to reduce variant classification inequities in underrepresented populations
Abstract
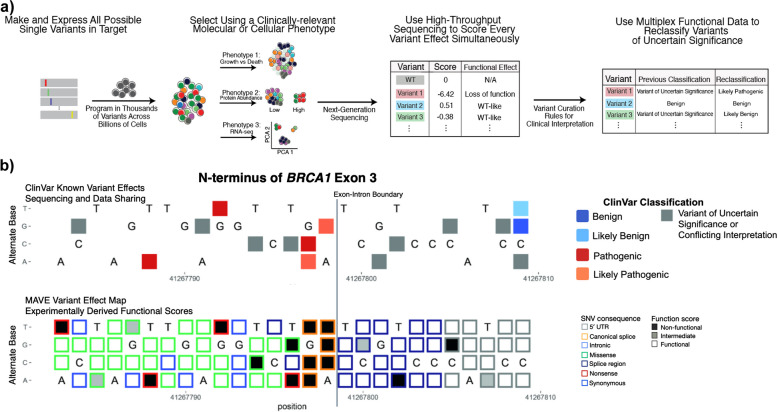
Background: Multiplexed Assays of Variant Effects (MAVEs) can test all possible single variants in a gene of interest. The resulting saturation-style functional data may help resolve variant classification disparities between populations, especially for Variants of Uncertain Significance (VUS).
Methods: We analyzed clinical significance classifications in 213,663 individuals of European-like genetic ancestry versus 206,975 individuals of non-European-like genetic ancestry from All of Us and the Genome Aggregation Database. Then, we incorporated clinically calibrated MAVE data into the Clinical Genome Resource's Variant Curation Expert Panel rules to automate VUS reclassification for BRCA1, TP53, and PTEN.
Results: Using two orthogonal statistical approaches, we show a higher prevalence (p ≤ 5.95e - 06) of VUS in individuals of non-European-like genetic ancestry across all medical specialties assessed in all three databases. Further, in the non-European-like genetic ancestry group, higher rates of Benign or Likely Benign and variants with no clinical designation (p ≤ 2.5e - 05) were found across many medical specialties, whereas Pathogenic or Likely Pathogenic assignments were increased in individuals of European-like genetic ancestry (p ≤ 2.5e - 05). Using MAVE data, we reclassified VUS in individuals of non-European-like genetic ancestry at a significantly higher rate in comparison to reclassified VUS from European-like genetic ancestry (p = 9.1e - 03) effectively compensating for the VUS disparity. Further, essential code analysis showed equitable impact of MAVE evidence codes but inequitable impact of allele frequency (p = 7.47e - 06) and computational predictor (p = 6.92e - 05) evidence codes for individuals of non-European-like genetic ancestry.
Conclusions: Generation of saturation-style MAVE data should be a priority to reduce VUS disparities and produce equitable training data for future computational predictors.
Keywords: All of Us; Benign; Equity; Genetic ancestry; Inequity; MAVE; Missense; Multiplexed assay of variant effects; Pathogenic; VUS; Variants of uncertain significance; gnomAD.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. JRL serves on the Scientific Advisory Board of Baylor Genetics. EV, JRL, and RAG declare that Baylor Genetics is a Baylor College of Medicine affiliate that derives revenue from genetic testing. BCM and Miraca Holdings have formed a joint venture with shared ownership and governance of Baylor Genetics which performs clinical microarray analysis and other genomic studies (exome sequencing and whole genome sequencing) for patient and family care. EV is a co-founder of Codified Genomics, a provider of genetic interpretation. The remaining authors declare that they do not have any competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
