

Molecular and Clinical Characterization of a Founder Mutation Causing G6PC3 Deficiency
- PMID: 39630167
- PMCID: PMC11618172
- DOI: 10.1007/s10875-024-01836-0
Molecular and Clinical Characterization of a Founder Mutation Causing G6PC3 Deficiency
Abstract
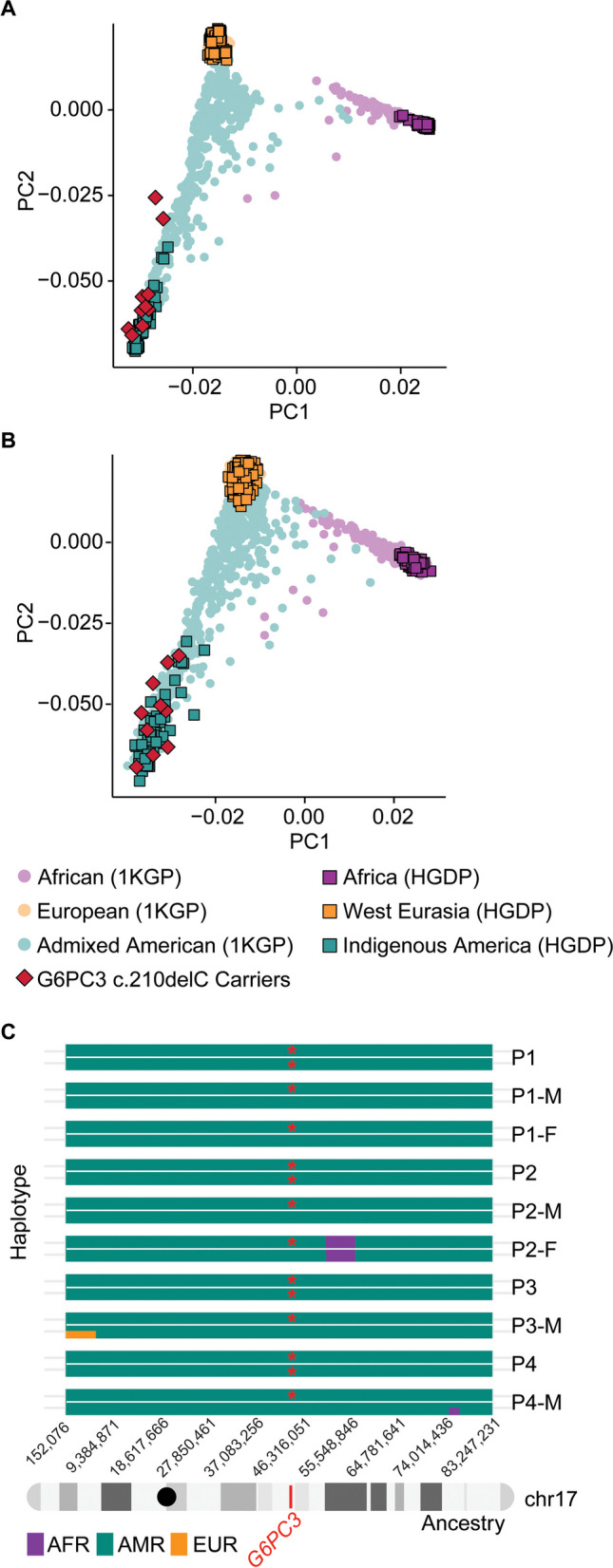
G6PC3 deficiency is a monogenic immunometabolic disorder that causes severe congenital neutropenia type 4. Patients display heterogeneous extra-hematological manifestations, contributing to delayed diagnosis. Here, we investigated the origin and functional consequence of the G6PC3 c.210delC variant found in patients of Mexican descent. Based on the shared haplotypes amongst mutation carriers, we estimated that this variant originated from a founder effect in a common ancestor. Furthermore, by ancestry analysis, we concluded that it appeared in the indigenous Mexican population. At the protein level, we showed that this frameshift mutation leads to an aberrant protein expression in overexpression and patient-derived Epstein-Barr Virus-immortalized B (EBV-B) cells. The neutropenia observed in G6PC3-deficient patients is driven by the intracellular accumulation of the metabolite 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) that inhibits glycolysis. We characterized how the c.210delC variant impacts glycolysis by performing extracellular flux assays on patient-derived EBV-B cells. When treated with 1,5-anhydroglucitol (1,5-AG), the precursor to 1,5-AG6P, patient cells exhibited markedly reduced engagement of glycolysis. Finally, we compared the clinical presentation of patients with the mutation c.210delC and all other G6PC3-deficient patients reported in the literature, and we found that the c.210delC carriers display all prominent clinical features observed in prior patients. In conclusion, G6PC3 c.210delC is a loss-of-function mutation that arose from a founder effect in the indigenous Mexican population. These findings may facilitate the diagnosis of additional patients in this geographical area. Moreover, the in vitro 1,5-AG-dependent functional assay used in our study could be employed to assess the pathogenicity of additional G6PC3 variants.
Keywords: Founder effect; G6PC3 deficiency; Inborn errors of immunity; Metabolic dysfunction; Primary immunodeficiency; Severe congenital neutropenia.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing Interests: The authors declare no competing interests.
Figures




Update of
-
Molecular and clinical characterization of a founder mutation causing G6PC3 deficiency.medRxiv [Preprint]. 2024 May 14:2024.05.13.24307299. doi: 10.1101/2024.05.13.24307299. medRxiv. 2024. Update in: J Clin Immunol. 2024 Dec 4;45(1):53. doi: 10.1007/s10875-024-01836-0. PMID: 38798393 Free PMC article. Updated. Preprint.
-
Molecular and clinical characterization of a founder mutation causing G6PC3 deficiency.Res Sq [Preprint]. 2024 Jul 11:rs.3.rs-4595246. doi: 10.21203/rs.3.rs-4595246/v1. Res Sq. 2024. Update in: J Clin Immunol. 2024 Dec 4;45(1):53. doi: 10.1007/s10875-024-01836-0. PMID: 39041036 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
