

Single chromatin fiber profiling and nucleosome position mapping in the human brain
- PMID: 39631398
- PMCID: PMC11704683
- DOI: 10.1016/j.crmeth.2024.100911
Single chromatin fiber profiling and nucleosome position mapping in the human brain
Abstract
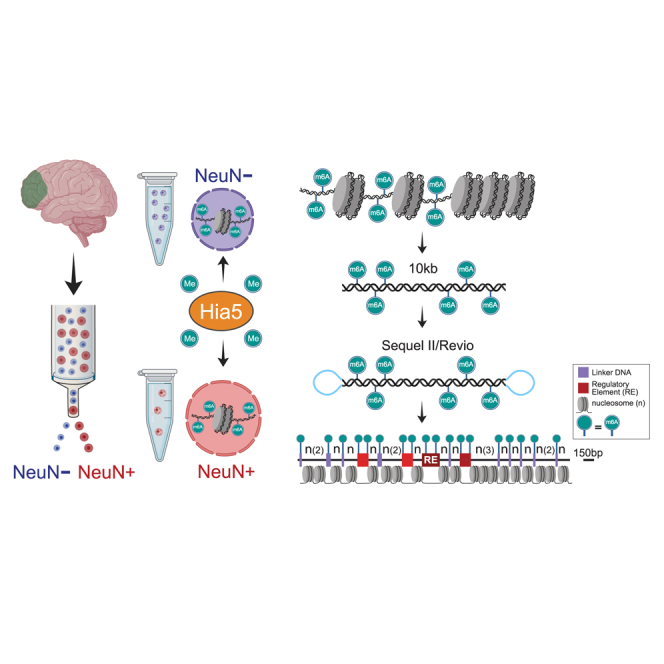
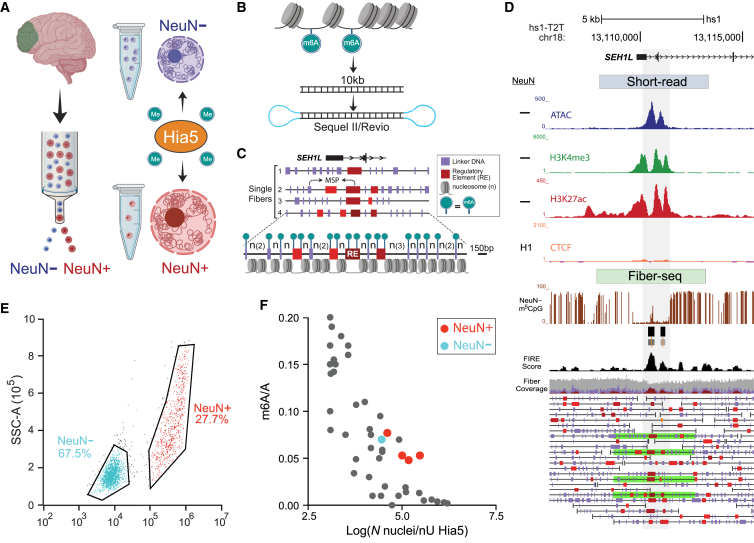
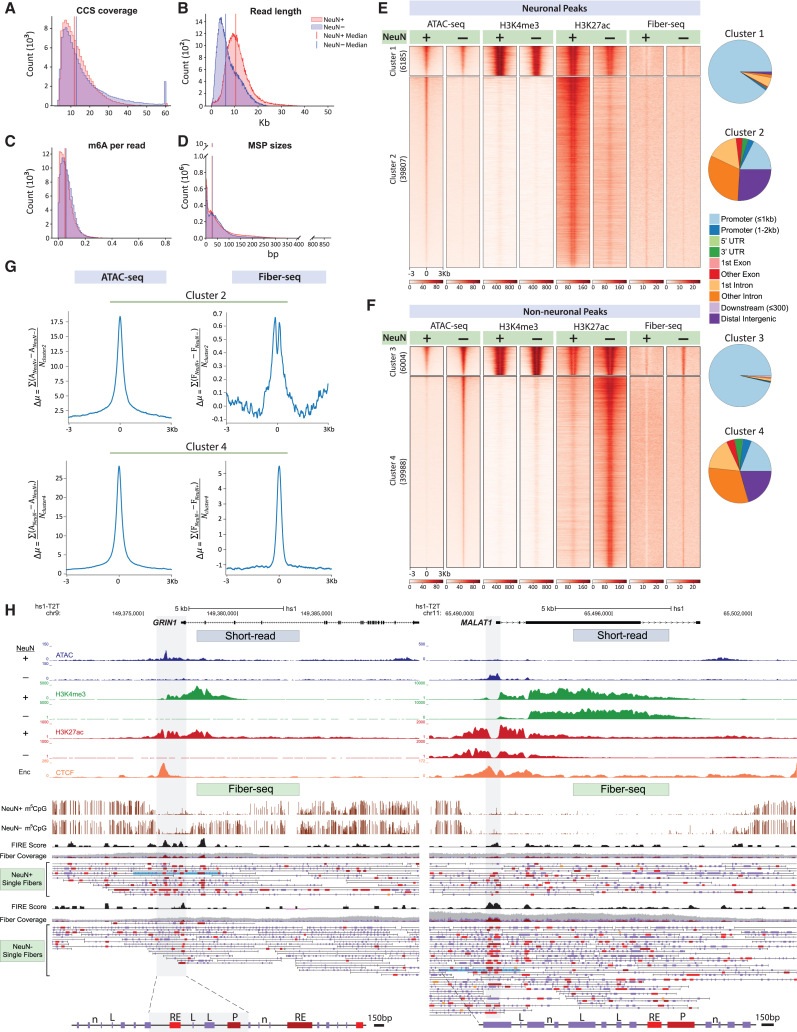
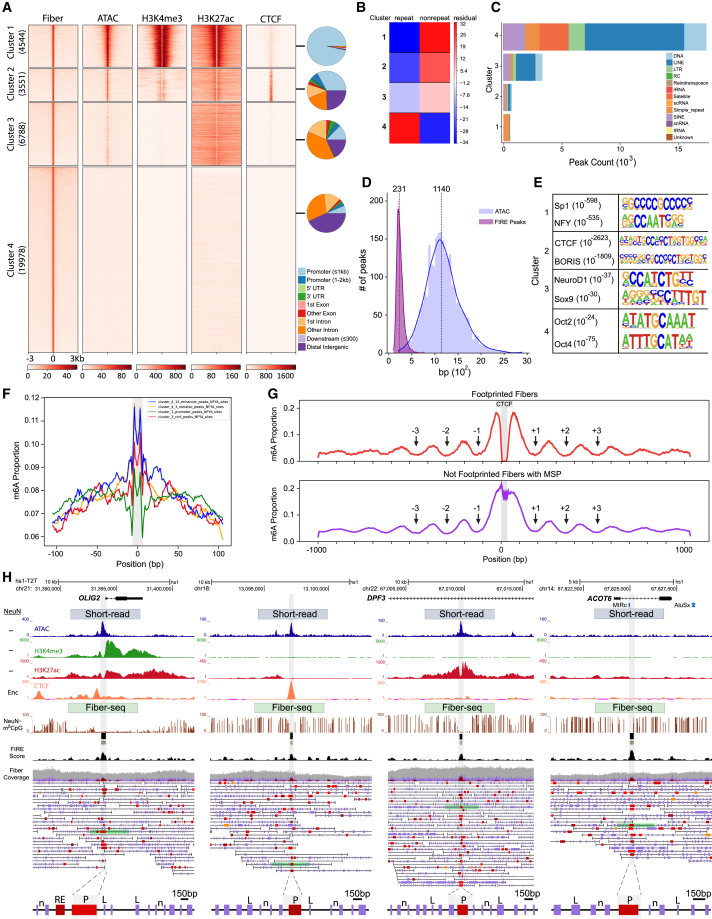
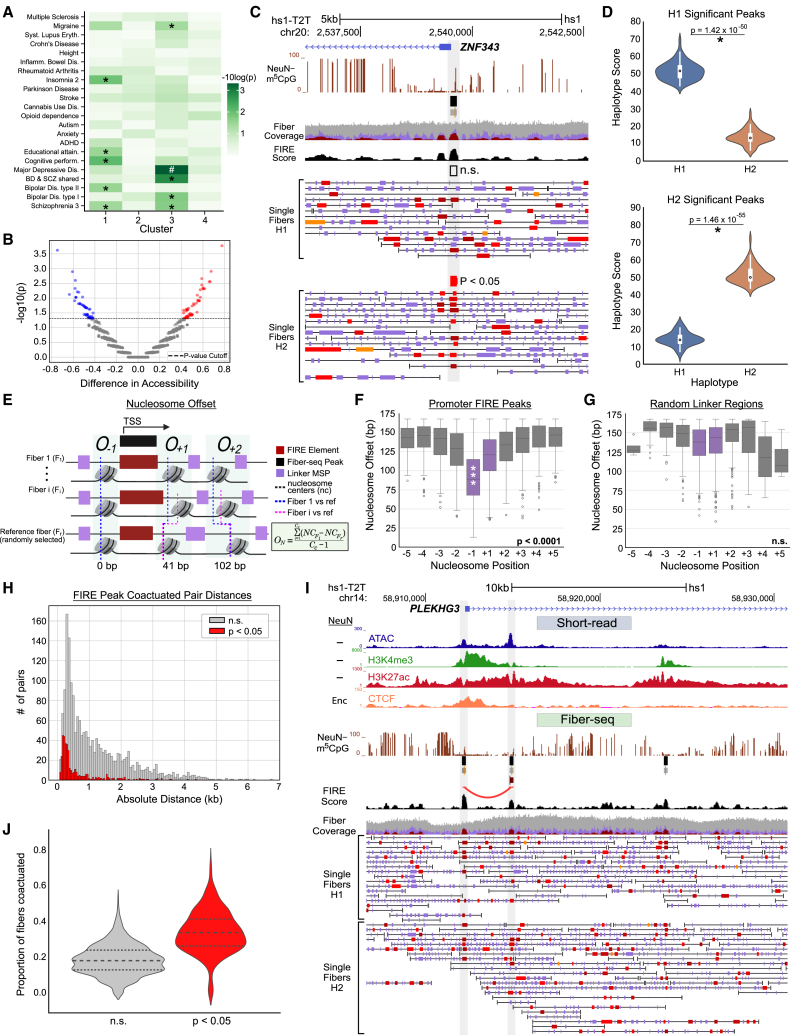
We apply a single-molecule chromatin fiber sequencing (Fiber-seq) protocol designed for amplification-free cell-type-specific mapping of the regulatory architecture at nucleosome resolution along extended ∼10-kb chromatin fibers to neuronal and non-neuronal nuclei sorted from human brain tissue. Specifically, application of this method enables the resolution of cell-selective promoter and enhancer architectures on single fibers, including transcription factor footprinting and position mapping, with sequence-specific fixation of nucleosome arrays flanking transcription start sites and regulatory motifs. We uncover haplotype-specific chromatin patterns, multiple regulatory elements cis-aligned on individual fibers, and accessible chromatin at 20,000 unique sites encompassing retrotransposons and other repeat sequences hitherto "unmappable" by short-read epigenomic sequencing. Overall, we show that Fiber-seq is applicable to human brain tissue, offering sharp demarcation of nucleosome-depleted regions at sites of open chromatin in conjunction with multi-kilobase nucleosomal positioning at single-fiber resolution on a genome-wide scale.
Keywords: CP: Biotechnology; CP: Neuroscience; Hia5 methyltransferase; adenine methylation; cytosine methylation; long-read sequencing; nucleosomal array; nucleosomal offset; postmortem brain; transcription factor footprint.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.B.S. is a co-inventor on a patent relating to the Fiber-seq method (US17/995,058).
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
