

CK1δ/ε-mediated TDP-43 phosphorylation contributes to early motor neuron disease toxicity in amyotrophic lateral sclerosis
- PMID: 39633494
- PMCID: PMC11619411
- DOI: 10.1186/s40478-024-01902-z
CK1δ/ε-mediated TDP-43 phosphorylation contributes to early motor neuron disease toxicity in amyotrophic lateral sclerosis
Abstract
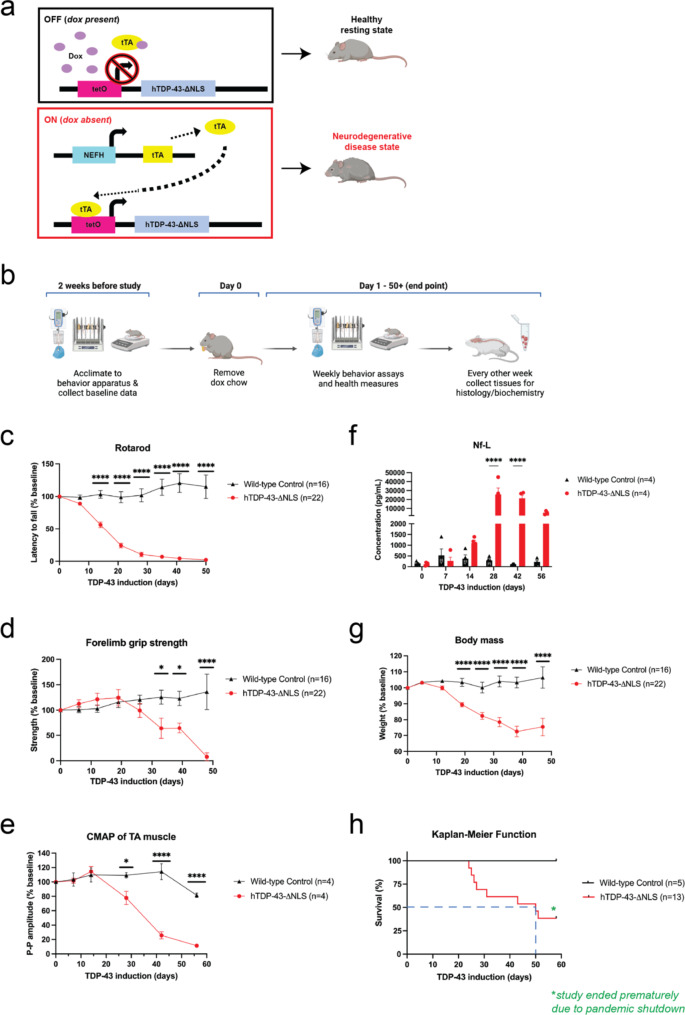
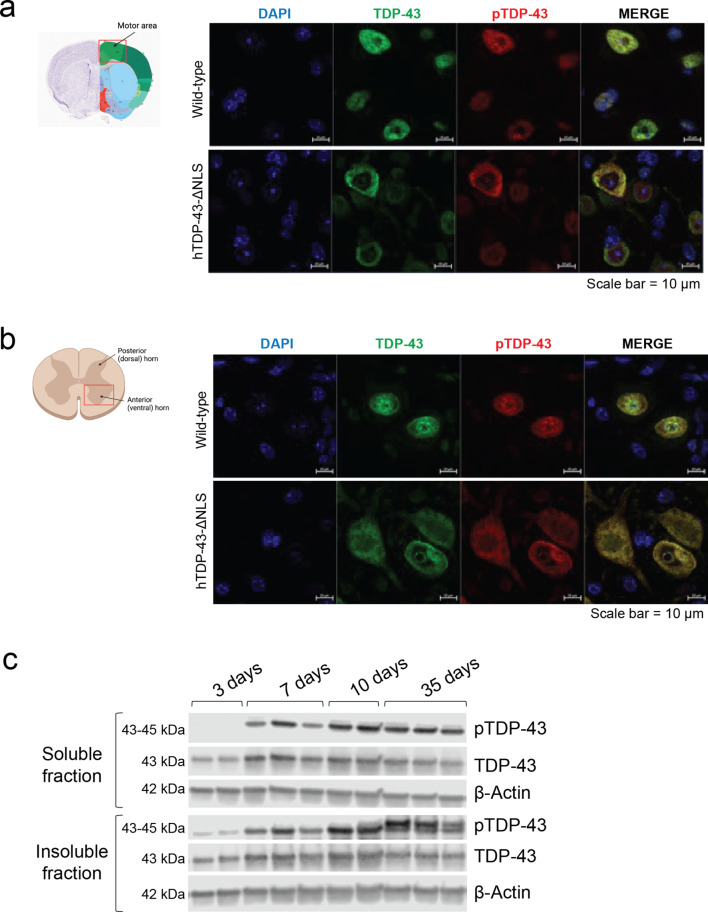
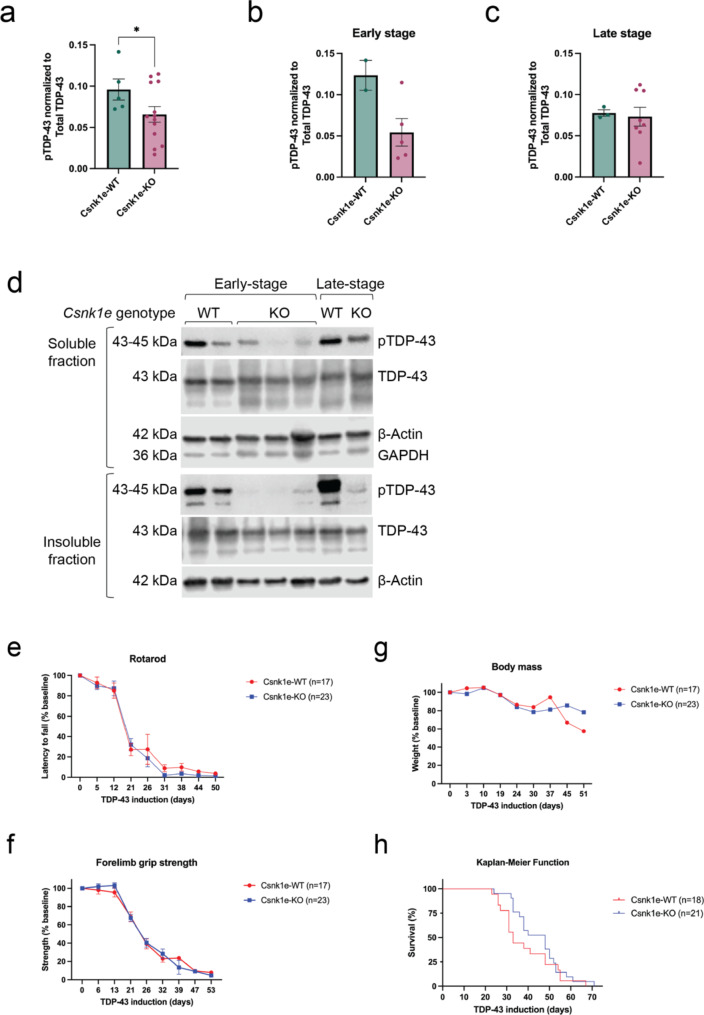
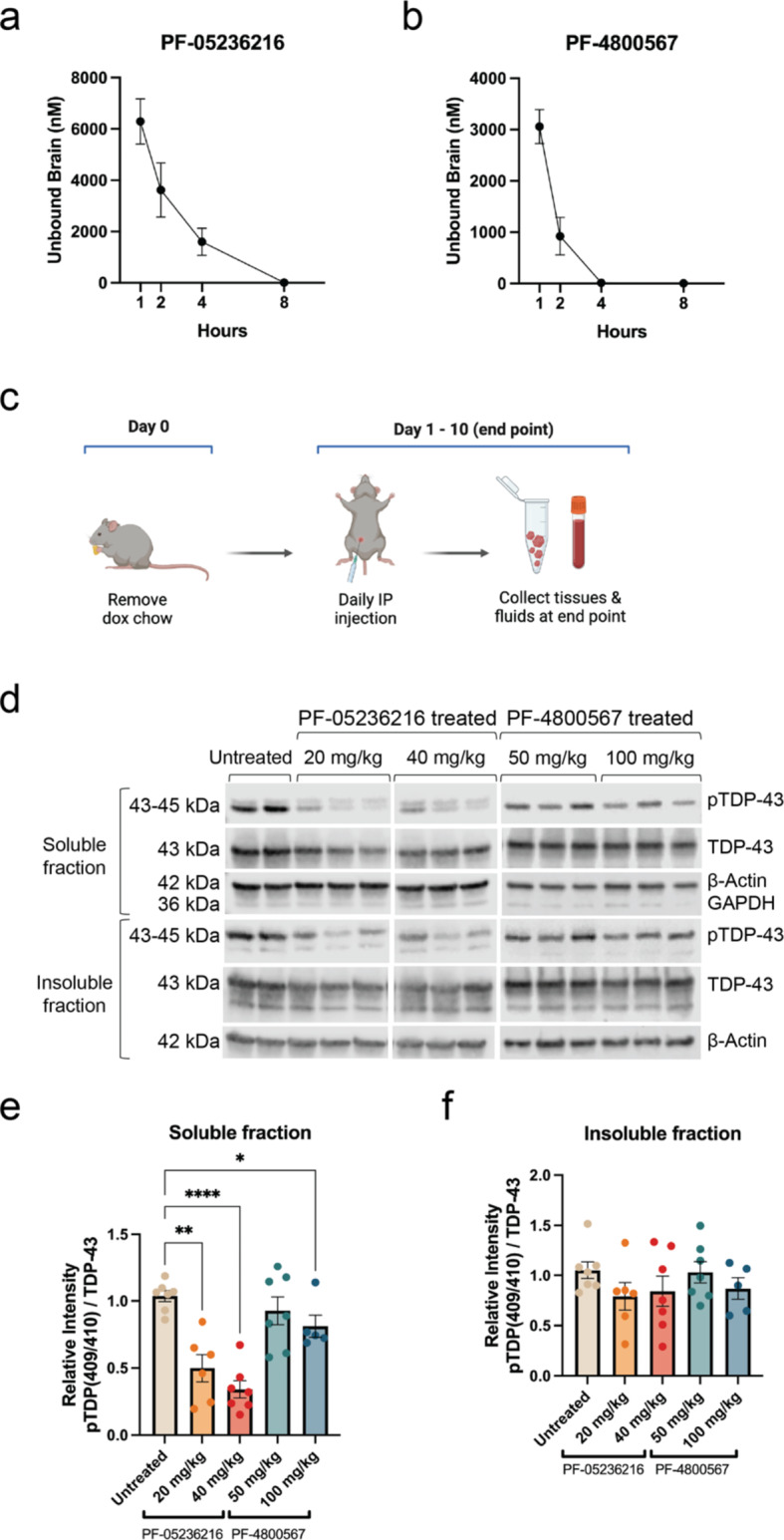
Hyperphosphorylated TDP-43 aggregates in the cytoplasm of motor neurons is a neuropathological signature of amyotrophic lateral sclerosis (ALS). These aggregates have been proposed to possess a toxic disease driving role in ALS pathogenesis and progression, however, the contribution of phosphorylation to TDP-43 aggregation and ALS disease mechanisms remains poorly understood. We've previously shown that CK1δ and CK1ε phosphorylate TDP-43 at disease relevant sites, and that genetic reduction and chemical inhibition could reduce phosphorylated TDP-43 (pTDP-43) levels in cellular models. In this study, we advanced our findings into the hTDP-43-ΔNLS in vivo mouse model of ALS and TDP-43 proteinopathy. This mouse model possesses robust disease-relevant features of ALS, including TDP-43 nuclear depletion, cytoplasmic pTDP-43 accumulation, motor behavior deficits, and shortened survival. We tested the effect of homozygous genetic deletion of Csnk1e in the hTDP-43-ΔNLS mouse model and observed a delay in the formation of pTDP-43 without significant ultimate rescue of TDP-43 proteinopathy or disease progression. Homozygous genetic deletion of Csnk1d is lethal in mice, and we were unable to test the role of CK1δ alone. We then targeted both CK1δ and CK1ε kinases by way of CK1δ/ε-selective PF-05236216 inhibitor in the hTDP-43-ΔNLS mouse model, reasoning that inhibiting CK1ε alone would be insufficient as shown by our Csnk1e knockout mouse model study. Treated mice demonstrated reduced TDP-43 phosphorylation, lowered Nf-L levels, and improved survival in the intermediate stages. The soluble TDP-43 may have been more amenable to the inhibitor treatment than insoluble TDP-43. However, the treatments did not result in improved functional measurements or in overall survival. Our results demonstrate that phosphorylation contributes to neuronal toxicity and suggest CK1δ/ε inhibition in combination with other therapies targeting TDP-43 pathology could potentially provide therapeutic benefit in ALS.
Keywords: Amyotrophic lateral sclerosis; Casein kinase 1 delta; Casein kinase 1 epsilon; Kinase inhibitors; Phosphorylation; TAR DNA-binding protein (TDP-43).
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All procedures were formed in accordance with NIH Guide for the Care and Use of Experimental Animals and Studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego. Consent for publication: N/A. Competing interests: The authors declare no competing interests.
Figures
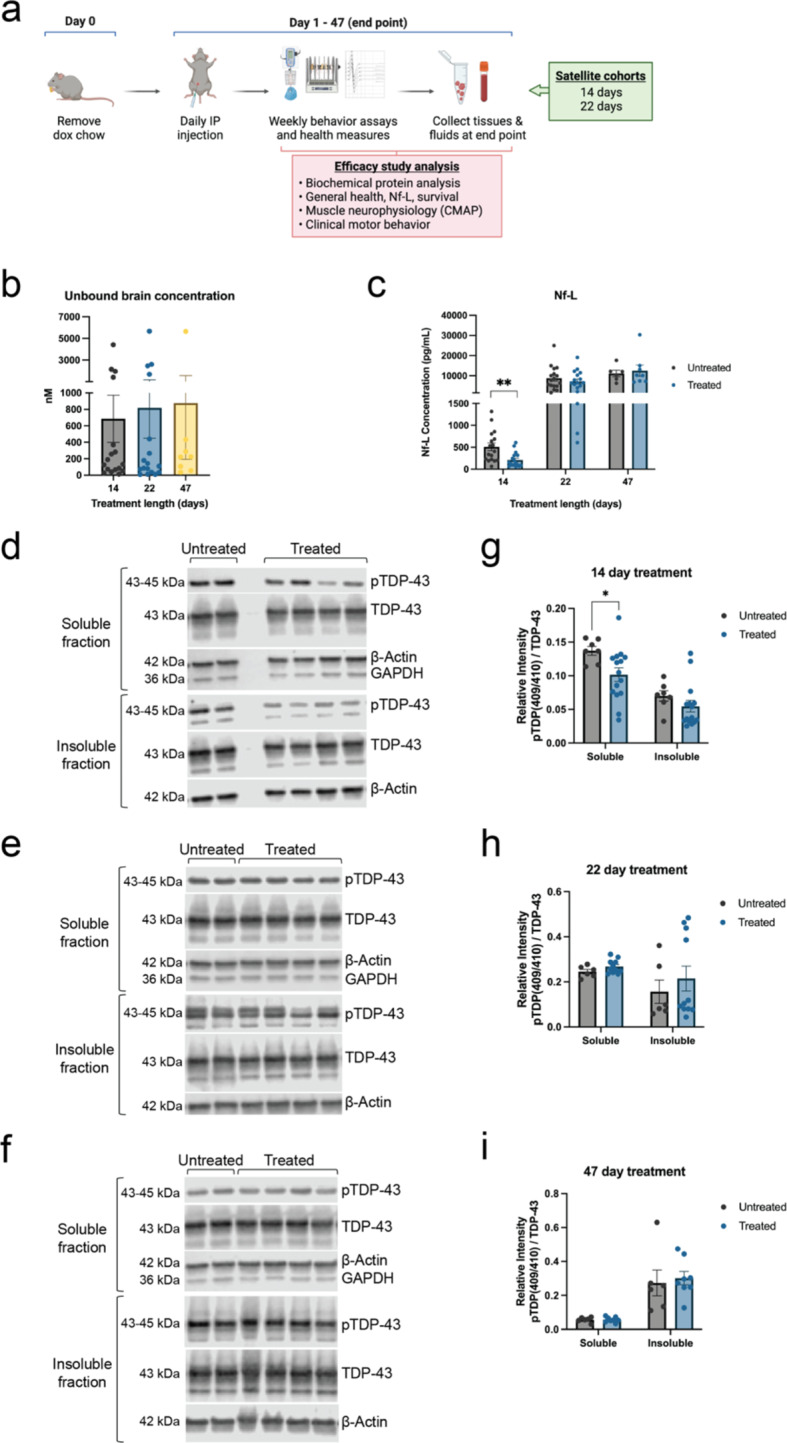
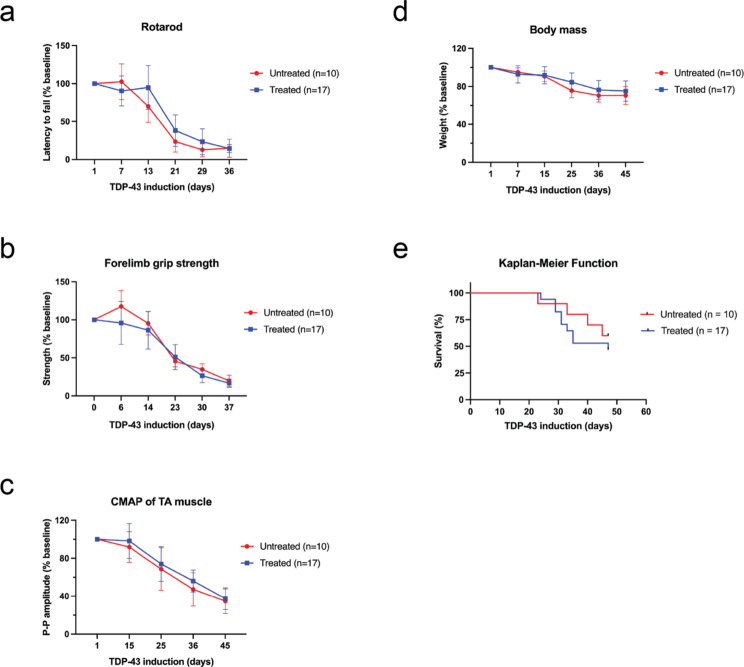
References
-
- Afroz T, Chevalier E, Audrain M, Dumayne C, Ziehm T, Moser R, Egesipe A-L, Mottier L, Ratnam M, Neumann M, Havas D, Ollier R, Piorkowska K, Chauhan M, Silva AB, Thapa S, Stöhr J, Bavdek A, Eligert V, Adolfsson O, Nelson PT, Porta S, Lee VM-Y, Pfeifer A, Kosco-Vilbois M, Seredenina T (2023) Immunotherapy targeting the C-terminal domain of TDP-43 decreases neuropathology and confers neuroprotection in mouse models of ALS/FTD. Neurobiol Dis 179:106050. 10.1016/j.nbd.2023.106050 - PubMed
-
- Alquezar C, Salado IG, De La Encarnación A, Pérez DI, Moreno F, Gil C, De Munain AL, Martínez A, Martín-Requero Á (2016) Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol Neurodegener 11:36. 10.1186/s13024-016-0102-7 - PMC - PubMed
-
- Benatar M, Zhang L, Wang L, Granit V, Statland J, Barohn R, Swenson A, Ravits J, Jackson C, Burns TM, Trivedi J, Pioro EP, Caress J, Katz J, McCauley JL, Rademakers R, Malaspina A, Ostrow LW, Wuu J, Hussain S, Cooley A, Li Y, Wallace M, Steele J, Hernandez JP, Medina J, Paredes ME, Manso A, Ravelo N, Levy W, Whitehead P, Zuchner S, Pasnoor M, Jawdat O, Jabari D, Farmakidis C, Glenn M, Dimachkie MM, Herbelin L, Tanui H, Anderson S, Walker M, Liu T, McCally A, Heim A, Currence M, Harness Y, Sieren J, Gibson E, Gutierrez G, Bussey D, Previte R, Kittrell P, Joshi A, Conger A, Hastings D, Caristo I, Marandi M, Carty S, Taylor JP, Wu G, Rampersaud E, Schule R, Blitterswijk MV (2020) Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology 95. 10.1212/WNL.0000000000009559 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
