

The FGFR inhibitor Rogaratinib reduces microglia reactivity and synaptic loss in TBI
- PMID: 39635532
- PMCID: PMC11614719
- DOI: 10.3389/fimmu.2024.1443940
The FGFR inhibitor Rogaratinib reduces microglia reactivity and synaptic loss in TBI
Abstract
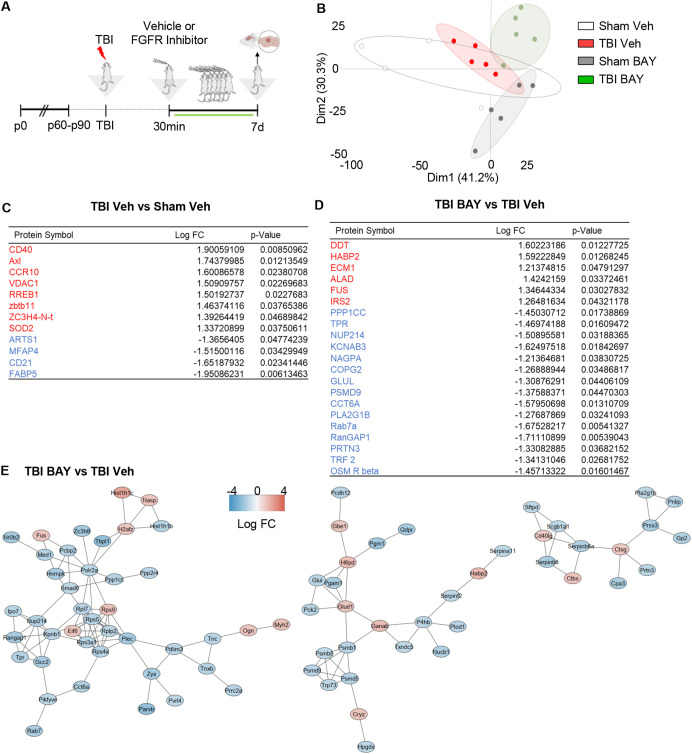
Background: Traumatic brain injury (TBI) induces an acute reactive state of microglia, which contribute to secondary injury processes through phagocytic activity and release of cytokines. Several receptor tyrosine kinases (RTK) are activated in microglia upon TBI, and their blockade may reduce the acute inflammation and decrease the secondary loss of neurons; thus, RTKs are potential therapeutic targets. We have previously demonstrated that several members of the Fibroblast Growth Factor Receptor (FGFR) family are transiently phosporylated upon TBI; the availability for drug repurposing of FGFR inhibitors makes worthwhile the elucidation of the role of FGFR in the acute phases of the response to TBI and the effect of FGFR inhibition.
Methods: A closed, blunt, weight-drop mild TBI protocol was employed. The pan-FGFR inhibitor Rogaratinib was administered to mice 30min after the TBI and daily up to 7 days post injury. Phosphor-RTK Arrays and proteomic antibody arrays were used to determine target engagement and large-scale impact of the FGFR inhibitor. pFGFR1 and pFGFR3 immunostaining were employed for validation. As outcome parameters of the TBI injury immunostainings for NeuN, VGLUT1, VGAT at 7dpi were considered.
Results: Inhibition of FGFR during TBI restricted phosphorylation of FGFR1, FGFR3, FGFR4 and ErbB4. Phosphorylation of FGFR1 and FGFR3 during TBI was traced back to Iba1+ microglia. Rogaratinib substantially dowregulated the proteomic signature of the neuroimmunological response to trauma, including the expression of CD40L, CXCR3, CCL4, CCR4, ILR6, MMP3 and OPG. Prolonged Rogaratinib treatment reduced neuronal loss upon TBI and prevented the loss of excitatory (vGLUT+) synapses.
Conclusion: The FGFR family is involved in the early induction of reactive microglia in TBI. FGFR inhibition selectively prevented FGFR phosphorylation in the microglia, dampened the overall neuroimmunological response and enhanced the preservation of neuronal and synaptic integrity. Thus, FGFR inhibitors may be relevant targets for drug repurposing aimed at modulating microglial reactivity in TBI.
Keywords: proteomics; reactive microglia; receptor tyrosine kinase; synapses; traumatic brain injury.
Copyright © 2024 Rehman, Froehlich, olde Heuvel, Elsayed, Boeckers, Huber-Lang, Morganti-Kossmann and Roselli.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures




References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous