

Rearrangements of viral and human genomes at human papillomavirus integration events and their allele-specific impacts on cancer genome regulation
- PMID: 39638560
- PMCID: PMC12047271
- DOI: 10.1101/gr.279041.124
Rearrangements of viral and human genomes at human papillomavirus integration events and their allele-specific impacts on cancer genome regulation
Abstract
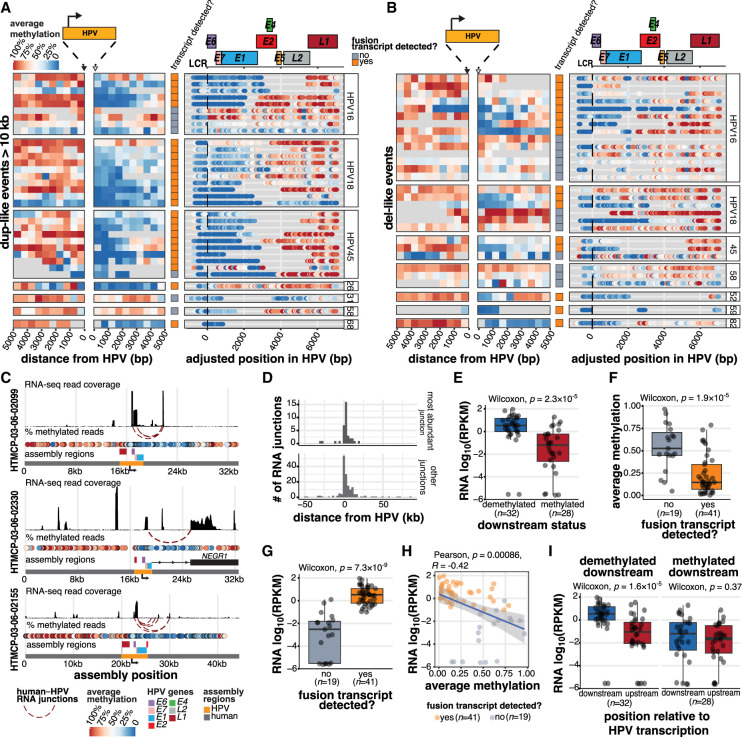
Human papillomavirus (HPV) integration has been implicated in transforming HPV infection into cancer. To resolve genome dysregulation associated with HPV integration, we performed Oxford Nanopore Technologies long-read sequencing on 72 cervical cancer genomes from a Ugandan data set that was previously characterized using short-read sequencing. We find recurrent structural rearrangement patterns at HPV integration events, which we categorize as del(etion)-like, dup(lication)-like, translocation, multi-breakpoint, or repeat region integrations. Integrations involving amplified HPV-human concatemers, particularly multi-breakpoint events, frequently harbor heterogeneous forms and copy numbers of the viral genome. Transcriptionally active integrants are characterized by unmethylated regions in both the viral and human genomes downstream from the viral transcription start site, resulting in HPV-human fusion transcripts. In contrast, integrants without evidence of expression lack consistent methylation patterns. Furthermore, whereas transcriptional dysregulation is limited to genes within 200 kb of an HPV integrant, dysregulation of the human epigenome in the form of allelic differentially methylated regions affects megabase expanses of the genome, irrespective of the integrant's transcriptional status. By elucidating the structural, epigenetic, and allele-specific impacts of HPV integration, we provide insight into the role of integrated HPV in cervical cancer.
© 2025 Porter et al.; Published by Cold Spring Harbor Laboratory Press.
Figures





Update of
-
Genomic structures and regulation patterns at HPV integration sites in cervical cancer.bioRxiv [Preprint]. 2023 Nov 5:2023.11.04.564800. doi: 10.1101/2023.11.04.564800. bioRxiv. 2023. Update in: Genome Res. 2025 Apr 14;35(4):653-670. doi: 10.1101/gr.279041.124. PMID: 37961641 Free PMC article. Updated. Preprint.
References
-
- Akagi K, Symer DE, Mahmoud M, Jiang B, Goodwin S, Wangsa D, Li Z, Xiao W, Dunn JD, Ried T, et al. 2023. Intratumoral heterogeneity and clonal evolution induced by HPV integration. Cancer Discov 13: 910–927. 10.1158/2159-8290.CD-22-0900 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical