

Discovery of BBO-8520, a First-In-Class Direct and Covalent Dual Inhibitor of GTP-Bound (ON) and GDP-Bound (OFF) KRASG12C
- PMID: 39642212
- PMCID: PMC11873722
- DOI: 10.1158/2159-8290.CD-24-0840
Discovery of BBO-8520, a First-In-Class Direct and Covalent Dual Inhibitor of GTP-Bound (ON) and GDP-Bound (OFF) KRASG12C
Abstract
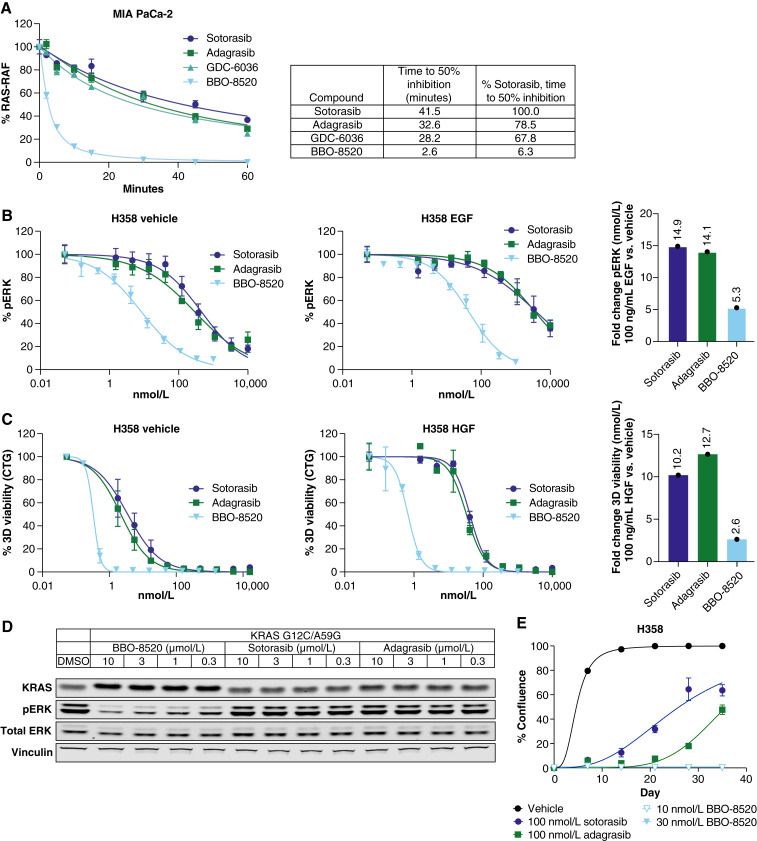
Approved inhibitors of KRASG12C prevent oncogenic activation by sequestering the inactive, GDP-bound (OFF) form rather than directly binding and inhibiting the active, GTP-bound (ON) form. This approach provides no direct target coverage of the active protein. Expectedly, adaptive resistance to KRASG12C (OFF)-only inhibitors is observed in association with increased expression and activity of KRASG12C(ON). To provide optimal KRASG12C target coverage, we have developed BBO-8520, a first-in-class, direct dual inhibitor of KRASG12C(ON) and (OFF) forms. BBO-8520 binds in the Switch-II/Helix3 pocket, covalently modifies the target cysteine, and disables effector binding to KRASG12C(ON). BBO-8520 exhibits potent signaling inhibition in growth factor-activated states, in which current (OFF)-only inhibitors demonstrate little measurable activity. In vivo, BBO-8520 demonstrates rapid target engagement and inhibition of signaling, resulting in durable tumor regression in multiple models, including those resistant to KRASG12C(OFF)-only inhibitors. BBO-8520 is in phase 1 clinical trials in patients with KRASG12C non-small cell lung cancer. Significance: BBO-8520 is a first-in-class direct, small molecule covalent dual inhibitor that engages KRASG12C in the active (ON) and inactive (OFF) conformations. BBO-8520 represents a novel mechanism of action that allows for optimal target coverage and delays the emergence of adaptive resistance seen with (OFF)-only inhibitors in the clinic. See related commentary by Zhou and Westover, p. 455.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
All BridgeBio Oncology Therapeutics (BBOT) authors are employees and stockholders of BBOT, a private company. B. Wang, R. Xiu, E. Wallace, Z. Zhang, and Y. Yang report a patent for PCT/US2022/037992(WO2023004102A2) pending. A.E. Maciag reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study, as well as a patent for PCT/US2022/037992(WO2023004102A2) pending, licensed, and with royalties paid from TheRas/BridgeBio. A.K. Sharma reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. A.H. Chan reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study, as well as a patent for PCT/US2022/037992 (WO2023004102A2) pending, licensed, and with royalties paid from TheRas/BridgeBio. M. Dyba reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study and owning stock in BridgeBio. B.P. Smith reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study, as well as owning stock in BridgeBio Pharma, Inc. D. Rabara reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. E.K. Larsen reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. J.-P. Denson reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. P.A. Alexander reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. M. Abreu Blanco reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. D.M. Turner reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study, as well as a patent for PCT/US2022/037992 (WO2023004102A2) pending, licensed, and with royalties paid from TheRas/BridgeBio. F.C. Lightstone reports a patent for PCT/US2022/037992(WO2023004102A2) pending and licensed to BBOT. K.-K. Wong reports grants from BridgeBio and Mirati during the conduct of the study, as well as other support from Cogent, Iambic, Pfizer, and Janssen outside the submitted work. A.G. Stephen reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. D.K. Simanshu reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study, as well as a patent for PCT/US2022/037992 (WO2023004102A2) pending, licensed, and with royalties paid from TheRas/BridgeBio. D.V. Nissley reports support from a Collaborative Research and Development Agreement with TheRas/BridgeBio and from NCI contract 75N91019D00024 during the conduct of the study. F. McCormick reports personal fees from BBOT and Leidos Biomedical during the conduct of the study, as well as personal fees from BridgeBio, Quanta, and Amgen outside the submitted work. No disclosures were reported by the other authors.
Figures






References
-
- Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRASG12C somatic mutations across race, sex, and cancer type. N Engl J Med 2021;384:185–7. - PubMed
-
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. . The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
